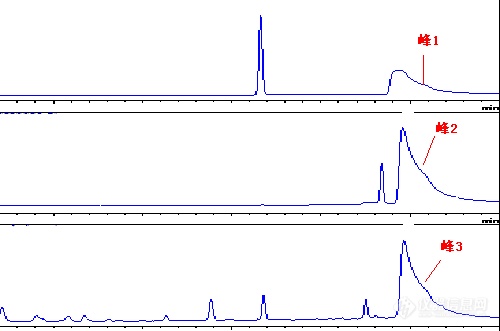
2、以下图谱中随着保留时间的延长,在图谱的末端都会出现一个看似“直角三角形”的峰,并且与进样物质无关,请您来解析。样品可能进样量超载,产生鬼峰和拖尾。建议把柱子重新冲洗,并且平衡好。
这个峰很高很锐 应该是梯度变化导致的吧
不是样品的 有可能是仪器某个位置的污染或柱子死保留导致的
可以走空白对照来辨别消除影响
可能水质量不好
我们现在用乐百氏的纯净水,杂质峰很明显,梯度洗脱时会有4个小峰出现。换屈臣氏的水就没有,但很贵啊。我们公司自己的milipore纯水机制的水也比乐百氏的强,但是不是自己部门的机器,用的不方便。
最大可能是梯度产生的
我也遇到过这种问题,不过是个倒着的峰.我们用的是剃度洗脱,我个人认为,可能与流动相比例瞬间改变有关.
我们做梯度洗脱的时候,经常这样。
有可能是使用的作为流动相的溶剂不纯
强保留物质一般会出现的峰型
我也觉得是流动相梯度洗脱导致的,可能是上一个梯度的累积,流速放慢一些会不会好一些
直接从柱子的清洗和流动相的置换上找原因
这个峰我最近就遇到过,按照
液相色谱故障的排除方法,从检测器开始排查一直到泵,都没发现问题,整的我买了跟新色谱柱,都没排除故障,整整花费了我三个月时间,最后发现是流动相受污染了。
还有一种可能是梯度变化太大,检测器出现的相应信号。
现在色谱纯乙腈很贵,买的国产的乙腈很多都产生鬼峰,郁闷好几次,要定的杂质都埋没在里面了
如果用的是剃度洗脱的话,这个很正常,可用空白进行判定。如果没有用洗脱,仍可逐一排除原因
前段时间用手动进样的时候也遇到这个问题
HPLC各部分都依次排查了
最后发现是流通阀有上次样品的残留
他们说应该是先插针,再打开进样器进样
与进的物质无关,如果梯度一样应该是梯度变化引起的
如果进样了空白溶剂,还有峰的出现,说明流动相污染概率大。如果更换流动相还出峰就说明是梯度原因了