
获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 xky0230699(xky0230699) 发表:有机合成液相色谱跟踪检测之我见
随着药典中高效液相色谱应用的增加,在有机合成跟踪检测上的作用也与日俱增,它的快速,灵敏,准确,可同时分析多组分等优点正符合跟踪需要。有机合成跟踪分析从严格性上来说,明显低于药典的相关要求(当然,成品分析是要求与药典和相关标准是一样的),检测的主要目的是反映实验的进程和副反应变化,为实验条件选择,优化提供依据。以下是本人对反相色谱法跟踪分析过程的一点总结。
一、 准备阶段
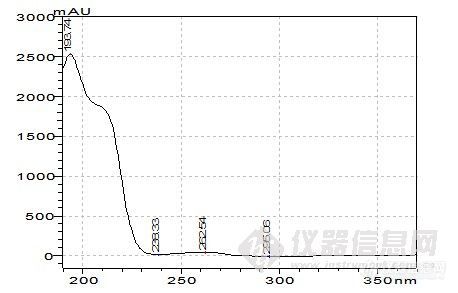
这一阶段的工作类似于方法开发过程。首先,要了解反应的底物(原料)和产物的结构,理化性质等,为流动相,PH,色谱柱的选择提供依据,(方法开发站内已有大量的帖子,这里不再重复说明),反应使用溶剂的性质,这个也很重要,因为溶剂经常PH是超出色谱柱的使用范围的(如胺类溶剂,酸性反应液等),且在检测条件下可能有吸收,会干扰或误导分析,要做空白溶剂对照,在不引起反应液改变的前提下,调节供试品溶液的PH。在能用反相色谱的情况下,尽量使用反相,操作简便,分析条件较容易摸索得到;第二,选择初始流动相,通常为80%甲醇-水,这一条件下,可以确定反应液所有组分的出峰时间,再以10%,5%或微调的比例调节有机相,从大到小,使之达到较佳的分离度(这一步如果有梯度泵,将带来极大的便利,几乎可以说是方法开发必备的利器)。检测波长低于220nm的,一般选择乙腈为有机相比较适合,但也要根据样品的溶解性加以综合考虑。根据峰形,分离度等,看是否需要加入调节剂,如缓冲盐(增加缓冲盐浓度常有延长保留时间的趋势,但不是很明显,当分离度在1.0-1.5之间时,或可加以运用),三乙胺,离子对试剂,四氢呋喃(作调节剂时比例常不超过10%),是否需要调节流动相的PH等,缓冲盐溶液通常为30mmonl/L的磷酸二氢钾溶液,扫尾剂常为0.5%或1%的三乙胺,或可增加两者的浓度。第三,波长的选择,由于反应液组分的复杂性与纯品的单一性不同,一般少用紫外分光光度计进行波长扫描,因为你扫描出的是多组分叠加后的吸收曲线,所以有DAD检测器是最适合的,其次是停泵扫描(见http://www.instrument.com.cn/bbs/shtml/20081011/1527048/)。我手上使用的是岛津SPD-20A的检测器,所以最常用的就是停泵扫描,此检测器有双波长检测功能,因此在第一次检测时,波长一般选择210nm(甲醇的截止波长)和254nm进行检测,此双波长下基本可以反映反应液的所有组分,确定各组分的出峰时间后,对各组分进行停泵扫描。最少扫描3次(曾遇上过假的扫描图谱),同时把扫描的吸收强度与相同时样量进色谱峰峰高对照,两者应基本一致,说明扫描正确。第四,柱温,我一般选择30℃,有需要再加以调整。
总而言之,这一阶段的最低要求是,要尽量使所有组分均被检出,各组分比例接近于实际含量,且达到一定的分离度和柱效;其次,是峰对称性在0.8-1.5之间即可;再次,就是要在尽量短的时间内完成检测以适应跟踪需求。尽可能先了解整个合成路线,使开发的方法能适用于整个合成过程,避免采用多个分析方法,以致每次都要对相应色谱峰进行定位和不断更换流动相,平衡色谱柱造成的时间浪费。
二、 跟踪阶段
此阶段检测重点是观察并如实记录反应液的变化情况,如各组分峰面积的变化,组分数目的变化,保留时间的变化及其与反应条件的关系。
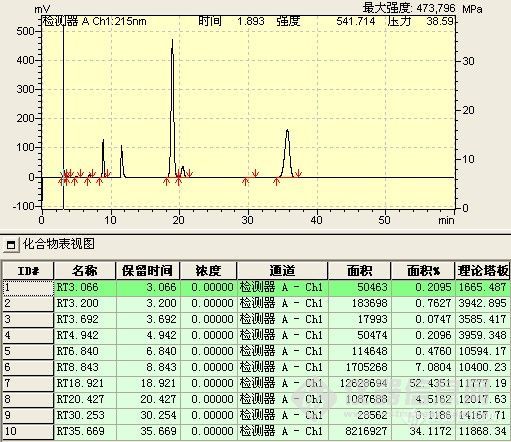
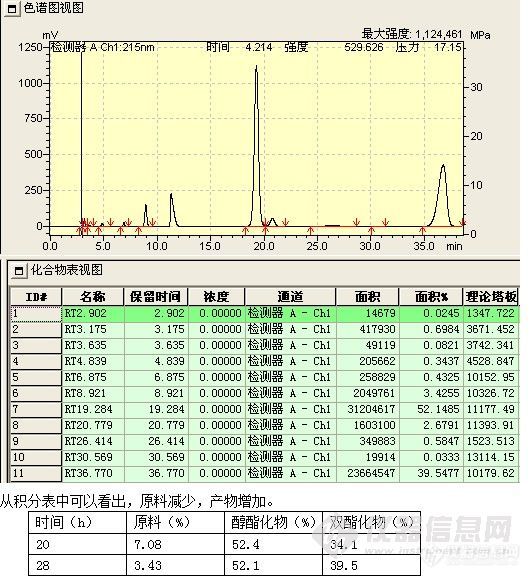
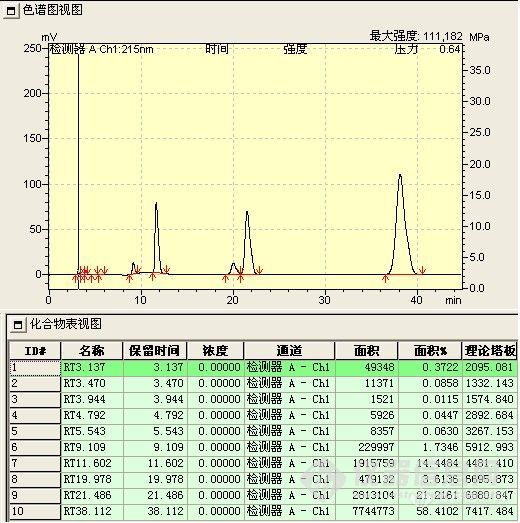
1、 峰面积的变化 正常情况下,应该原料峰面积减少,而产物峰面积增加。首先,原料峰的变化最容易判断,通过原料的保留时间定性和峰面积变化,就可判断原料是否有反应。其次,产物的峰面积应增加,但峰面积增加的未必就是所要的产物,也可能是副产物或中间态。可以通过以下判断:(1)最直观的表现,原料反应完全后,新出现的最大的峰往往就是产物峰;(2)反应机理 通过机理可能判断副反应进行的难易程度对峰面积的大小进行对照,如副反应很难进行,而有一色谱峰随着原料的减少而明显增加,那可能就是产物;(3)物质的极性 根据反应前后极性的变化,与原料进行比较,判断产物峰的可能位置,同时判断副产物的极性加以对照,并结合硅胶TLC的点位判断;(4)根据原料上引入基团是助色团还是减色团,与在双波长下峰面积不同的比较。(5)在原料基本反应完全后,提取,结晶出产物,以熔点,官能团鉴别来再一次确认;(7)如果有下一步反应,取产物进行下一步反应,看反应现象是否符合理论,符合,则确定反应产物正确。(7)以上方法都无法判断,可选择红外,质谱,核磁等进一步鉴别。产物确认后,以结晶后的产物返回对色谱峰进行定位。













































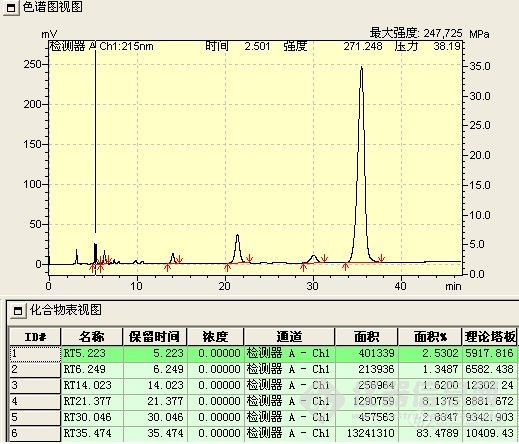
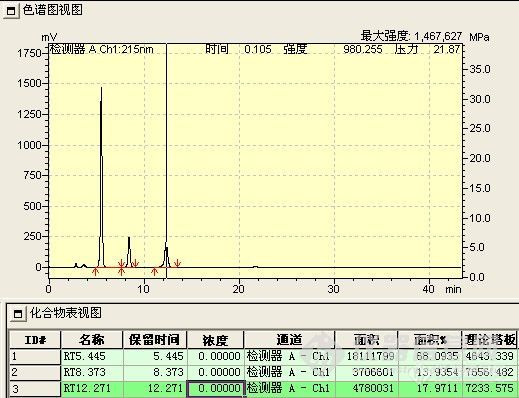
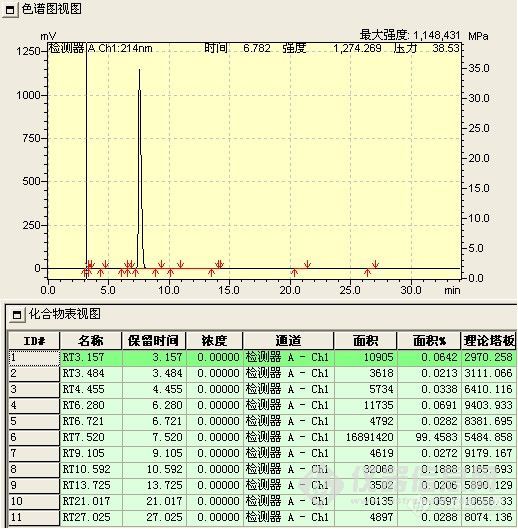

 好好学习,谢谢楼主分享
好好学习,谢谢楼主分享
