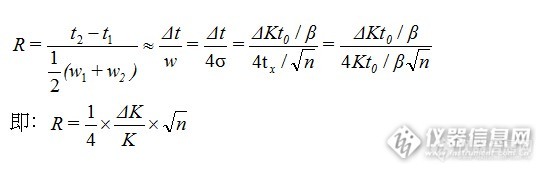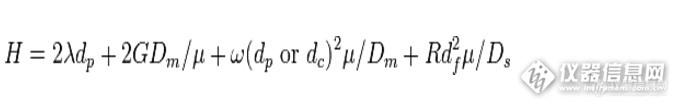原因色谱分离度又称分辨率,用来表征两个相邻色谱峰的分离程度,用R表示。R等于相邻色谱峰保留时间之差与两色谱峰峰宽均值之比。计算公式为:
![]()
R越大,表明相邻两组分分离越好。一般说当R<1时,两峰有部分重叠;当R=1.0时,分离度可达98%;当R=1.5时,分离度可达99.7%。通常用R=1.5作为相邻两组分已完全分离的标志。当R=1时,称为4σ分离,两峰基本分离,裸露峰面积为95.4%,内侧峰基重叠约2%。R=1.5时,称为6σ分离,裸露峰面积为99.7%。R≥1.5称为完全分离。《中国药典》规定R应大于1.5。为了判断分离物质对色谱柱在色谱柱中的分离情况,常用分离度作为柱的总分离效能指标,
从塔板理论可以推论出:
![]()
从这里我们可以清晰地看到影响分离度R的因素只有三个,即:两临近组分的分配系数(容量因子)之差ΔK、平均分配系数K、柱理论塔板数n。因此分离度R与柱长L(或柱理论塔板数n)的平方根成正比,与分配系数之差ΔK成正比,与平均分配系数K成反比。分配系数与固定相的性质、厚度及柱温有关:①与样品极性相似的固定相,分配系数大;②固定液液膜厚度增加,分配系数增大;③温度降低,分配系数增加。
因此我们仅从塔板理论来看,载气流速并不影响分离度R的。我们知道,这并不符合我们的实际经验。事实上,分离度R与载气流速是密切相关的。为什么会出现这个情况呢?这是因为塔板理论的四个假设并不成立,存在偏差。为了修正这些偏差,1956年荷兰人Van Deemter提出了速率理论
。
速率理论是对塔板理论的一个修正。范第姆特方程(Van Deemter equation)在色谱学中是综合考虑了分离过程中引起峰展宽的物理因素、动力学因素和热力学因素后得到的单位柱长的总峰展宽与流动相流速的关系式。
一般来说,影响峰展宽的因素包括多路径效应,扩散(纵向和横向)与固定相和流动相间的传质阻力。通常用理论塔板高度来表示色谱分离过程中的峰展宽。范德姆特方程呈双曲形函数的形式,表明流动相的流速存在一个最优值,在该点柱效最高。范第姆特方程最常用的形式如下式所示,该式直观地反映了流动相流速对于分离的影响。
![]()
式中H 为理论塔板高度,A, B,C为常数,μ表示流动相的流速。
A项反映的是被分析物在填充柱中可能采取不同的路径,因而经过的路程也不一样长,引起色谱峰的展宽,这就是“多路径效应”。在毛细管开管柱中不存在多路径效应,这一项为零。
B/μ表示的是因为色谱柱各部分存在浓差而引起的纵向扩散带来的峰展宽。
Cμ表示达到固定相与流动相的平衡之后由于在固定相与流动相传质存在着阻力引起的峰展宽。
范第姆特方程的完整形式如下:
![]()
式中:
λ 是一个与柱内填料粒度均一性与填充状态有关的常数;
dp 为填料的粒径;
G、ω和R 为常数;
Dm 是被分析物在流动相中的分子扩散系数,与载气的性质和柱温有关;
dc 是毛细管的直径;
df 是固定相的膜厚度;
Ds 是被分析物在固定相中的分子扩散系数,与固定相的性质有关。
由以上相关理论可以推论出,色谱柱的分离度与色谱柱的长度、固定相的性质、液膜厚度、载气流速和性质、柱温均有关系。
①分离度R与柱长L的平方根成正比,因此,采用较长的色谱柱可以获得更高的分离度,但相应的分析时间也会延长。而且色谱柱越长压降越大,柱过长会增大进出口压力比,相反会降低分离度。
②固定相的性质与分析物越相似,分离度越高;
③从上述公式中可以看出,液膜厚度的增加会导致塔板高度的增加,柱理论塔板数降低,同时还会增加分析物在固定相中的分配系数,理应导致分离度降低,但事实上,在分配系数增加的同时,ΔK增加更为显著,所以液膜厚度的增加能提高分离度;
④分子质量大的载气(N
2、Ar)的优点是扩散作用小,缺点是在柱中压降大、流速慢,即分析周期长。分子量较小的载气(H
2、He)传质阻力小,有利于提高分析速度,但浓度较高的介质易在其间形成扩散,影响分离度,所以在实际测量中H
2、He气体一般都用于介质浓度较低的区域并提高其流速,减少扩散的影响。
⑤色谱柱存在一个最佳载气流速,但实际分析中为缩短分析时间,流速通常稍大于最佳流速。
⑥提高柱温能提高分析物在流动相和固定相中的扩散系数,降低传质阻力,加快分析速度,同时使色谱峰变窄变高,但会导致分离度的下降。
⑦此外,色谱分离是样品在固定相和流动相中反复分配的过程,过载后本来该进入固定相的样品在固定相上得不到保留(例如:本来在这点有50%的样品能保留在固定相上,现在只能保留30%,因为固定相上样品饱和了),就被流动相推着往前走,因此造成了保留变化(主要是分配系数发生了改变),因此进样量与分离度也是有关系的。
因此,引起分离度降低的原因可能有:a.载气流速设置不当;b.柱温设置不当;c.色谱柱受到污染,柱效下降;d.进样量太大;e.样品的浓度发生改变。
解决方案①载气流速设置不合理,可以通过降低载气流速来提高分离度;
②柱温设置不当,降低柱温,或改用程序升温;
③色谱柱受污染,应清洗柱子;
④减少进样量,提高进样技术;
⑤提高气化室温度;
⑥减少系统的死体积,主要是色谱柱要连接到位,毛细管色谱柱要分流,选择合适的分流比。
(4) 案例分析对
气相色谱而言,正常情况下,升温速度越大,出峰绝对时间差越小,分离度R会因此减小。但有时也会出现一些反常的情况,譬如下面这个例子:
![]()
苯胺和苯酚的检测,用二硫化碳萃取后直接进样,程序升温分离。发现随着升温速率的增大,二者分离度反而显著提高,似乎与前面的结论完全不符合。
实质上其原因是因为程序升温的起始温度设置过低,随着升温速度加快,峰拖尾明显好转,峰形变得更对称了,峰宽因此而减小,从而出现这种反常现象。根据速率理论,温度升高后,固定液黏度减小,这个时候如果原来存在过大的传质阻力,将会因此得到好转。过大传质阻力的典型现象就是峰拖尾严重,有点类似存在死体积或强吸附。
在实际检测中,到底应选用恒温分离还是程升分离,其实首先要明确程序升温的目的。程升多应用于组分沸程宽的混合物的分离的,简单说就是分析样品中各组分的沸点差异比较大的样品多用程序升温来做,如果不是这样的情况,不建议使用程序升温。
下面以一个例子的分离来说明恒温和程序升温对难分离物质对进行分离的影响,如图2所示,A的温度程序是240℃,恒温12min后20℃/min升温至300℃;B的温度程序是180℃起,10℃/min程序升温至300℃。
![]()
从图中对比可以看出,程序升温时大峰后面的两个小峰分离度较差,且和大峰的分离度也不是很好,经过较高的一个温度恒温后,那两个小峰的分离度得到了有效的改善,效果还是比较明显的。但这并不意味着恒温的分离度一定优于程序升温。
当依靠温度程序的优化无法从根本上改变分离度时,最有效的办法就是更换极性或者换根长柱子试试。