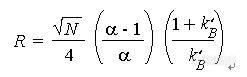0
0【线上讲座33期】液相色谱柱使用疑难问题解析主讲人:上海月旭公司技术总监 资深液相色谱应用专家 姚立新(仪器信息网id: Plexu)活动时间:2010年7月26日---8月7日
我们热烈欢迎plexu先生光临仪器论坛液相色谱版面进行讲座! ![]()
导言: Thomas Friedman说“世界是平的”,IT及其它科学领域的技术和工艺已经全球化而不再为欧美国家独占。随着色谱技术在全球医药、化工、生物、环保和食品行业的广泛性应用,色谱应用时遇到的技术疑难问题也已全球化了。
液相色谱柱是
液相色谱仪的心脏,承担着
液相色谱技术中最核心的分离功能,解决
液相色谱柱使用中的技术难题对提高
液相色谱应用的整体水平有着重要意义。
希望大家借此次交流机会,共同参与探索
液相色谱柱在使用中的任何问题,欢迎大家就
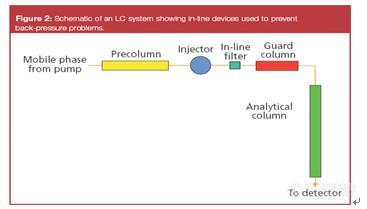
液相色谱柱的安装,使用和维护知识,以及各种柱压、峰形和色谱柱寿命等问题前来提问,也欢迎
液相色谱方面的高手前来与plexu交流切磋。
![]()
内容提纲:一、
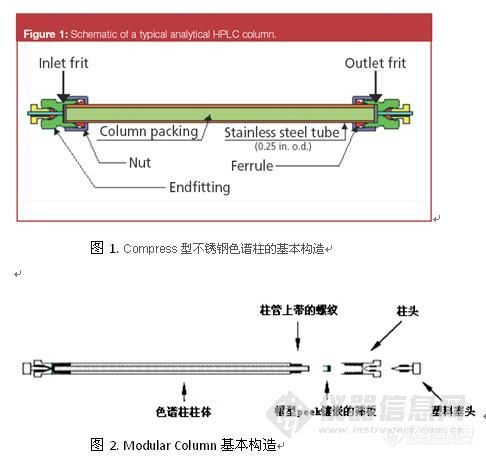
液相色谱柱的安装、启用和维护中的重点注意事项
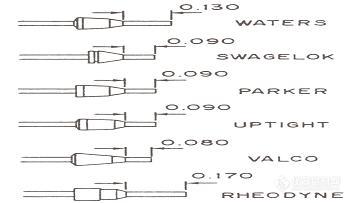
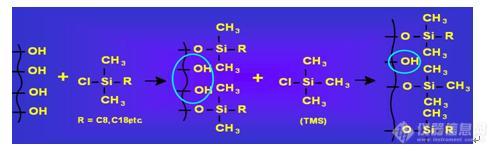
1、柱头类型和不锈钢毛细管接头的匹配
2、溶剂的匹配转换
3、新柱使用前的平衡和老化
4、pH使用范围
5、色谱柱的保存
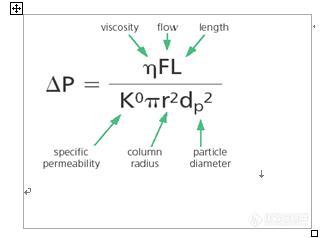
二、柱压问题
1、填料破碎和使用后,有填料粉末生成
2、颗粒物堵塞引起柱压上升和对策
1)预防措施
2)故障排除
3、化学污染物引起柱压上升和对策
1)预防措施
2)故障排除
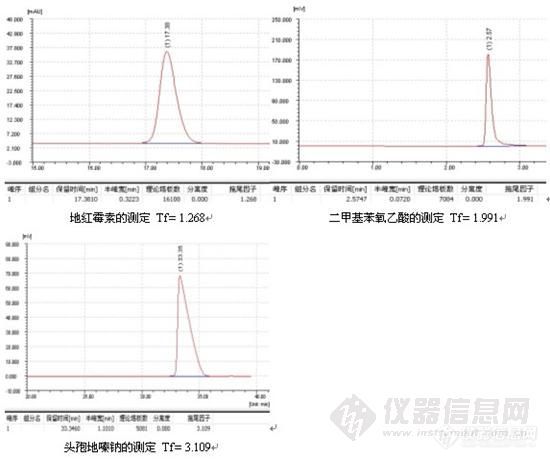
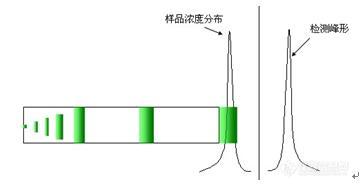
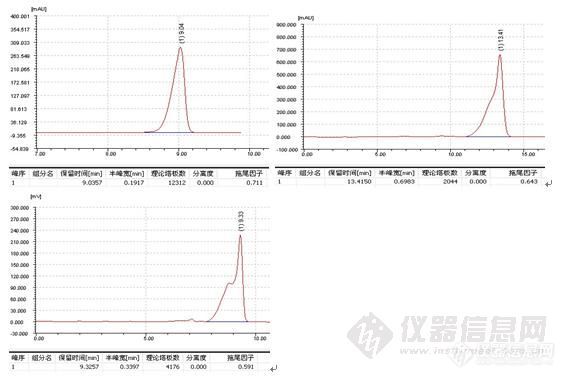
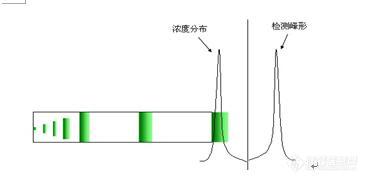
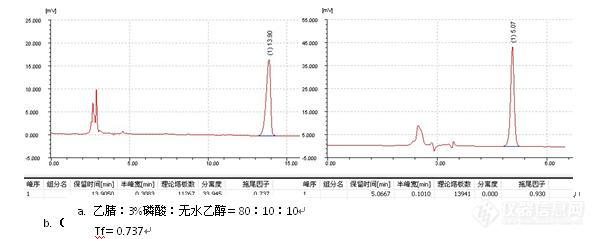
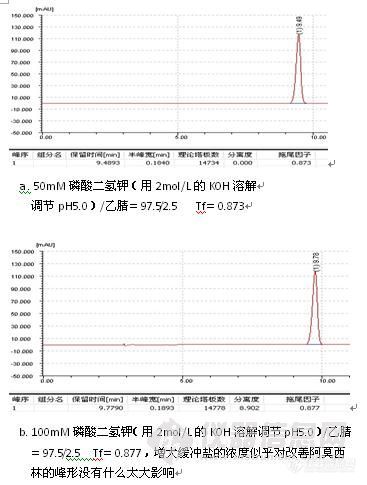
三、峰形问题
1、峰后拖
2、峰前延
3、 其他峰形问题
四、保留时间问题
1、 保留时间的重现性
2、 保留时间漂移
3、 柱与柱之间的重现性
4、 批与批之间的重现性
五、寿命问题
六、其他疑难的色谱技术问题
![]()
提问时间:2010年7月26日-2010年8月7日
答疑时间:2010年7月26日-2010年8月7日
特邀佳宾:液相色谱版面的版主以及
液相色谱界的专家
参与人员:全体注册用户
活动细则:1、 请大家就
液相色谱柱应用技术问题进行提问,直接回复本帖子即可,自即日起提问截至日期2010年8月7日
2、凡积极参与且有自己的观点的,或对常见的色谱柱应用问题进行提问的,都有机会获得月旭公司提供的丰厚奖品。
特设
积极参与奖20名 将获得月旭公司提供的
价值36元天堂伞、安利牙膏、实木双层鞋架(以上任选一)
![]()
![]()
![]() 精彩问答奖 3名
精彩问答奖 3名 将获得月旭公司提供的
价值200元以上飞利浦剃须刀或星空海龟一个
![]()
![]()
![]()
3、提问格式:
为了规范大家的提问格式,请按下面的规则来提问 :
plexu您好!我有以下问题想请教,
请问:……
![]()
说明:
本讲座内容仅用于个人学习,请勿用于商业用途,由此引发的法律纠纷本人概不负责。
虽然讲座的内容主要是对知识与经验的讲解、整理和总结,但是也凝聚着笔者大量心血,版权归月旭公司及plexu所有。
本讲座是根据笔者对资料的理解写的,理解片面、错误之处肯定是有,欢迎大家指正。
![]()