峰形后拖解决方案和实例
继前一篇《峰形前拖解决方案》,我们对峰形后拖的情况及其解决方案也进行了总结。与《峰形前拖解决方案》相同,在这里我们也同样的撇开所有的污染、塌陷、死体积等其它因素,假设系统是好的,柱子是新的、好的,单纯探讨
液相方法与峰形后拖之间的关系,通过调整
液相方法来解决峰形前拖的问题。这样有利于厘清拖尾产生的根本原因,对峰形拖尾的问题提供一些经验上解决方案。
首先让我们了解一下峰形后拖的几种常见形式,三种常见的拖尾如下:
![]()
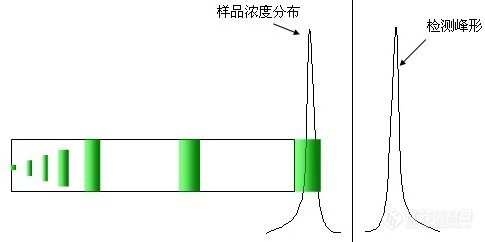
系统是好的,柱子是好的,为什么会产生后拖?我们还是回顾一下,色谱分离的一般过程,正常的峰形应该是样品在色谱柱上均匀前移的情况下得到的,浓度分布在整个通过色谱柱柱床的过程中任何时候都呈正态分布,如下图所示:
![]()
色谱分离是一个物理过程,样品分子经过在固定相与流动相之间的多次分配、吸附解吸,由于不同分子与固定相和流动相之间的相互作用力的差异而使它们在色谱柱中的移动速度不同,以此实现分离的目的。相同色谱条件下,有些化合物容易产生拖尾,有些则不产生拖尾,这说明拖尾的产生跟样品本身的性质是密切相关的,通常非极性和弱极性的化合物能获得良好的峰形,而带有-COOH、-NH2、-NHR、-NR2等极性基团的化合物则比较容易产生拖尾。
![]()
可以想象一下,样品分子在分析过程所处的环境,一是流动相,二是固定相,流动相可以自由流动的,均一性比较好,因此可以认为样品分子四周都被相同组成的流动相所包围,其对每个样品分子的相互作用力也是均匀的、对称的,不应该是引起浓度分布发生变化的主要原因。考虑固定相,固定相中除了C18长链外还有填料键合时残余的硅羟基,由于C18长链是非极性的,其与样品分子的相互作用力也是很微弱的范德华力,而硅羟基则具有一定的极性Si-Oδ-Hδ+,在pH一定的条件下甚至还会发生电离,形成-Si-O- 形式的离子态。Si-Oδ-Hδ+和-Si-O-对于极性化合物之间的作用力则是一种极性的静电作用力,这种作用力比范德华力要强得多,同时,因为硅胶表面键合了C18长链,由于空间位阻作用,样品分子中能接触到残余硅羟基的机会不会很多,只有少部分的分子才能与残余硅羟基产生作用。这样,大多数的样品分子如非极性分子一样均匀前移,而少量的样品分子由于这种静电作用力的存在而被推迟洗脱出来,导致样品分子的浓度分布发生变化,产生后拖。拖尾严重的程度与样品分子极性大小和残余硅羟基的多少有着直接的关系。
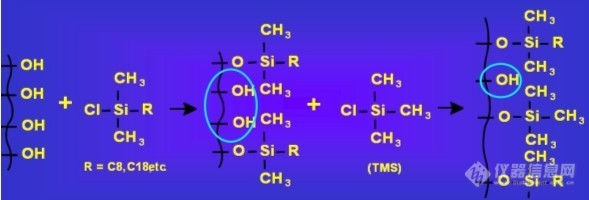
既然残余硅羟基对样品的次级保留效应是导致峰形后拖的主要原因,那我们先了解一下残余硅羟基是怎么回事。我们先借用一下wsy18的一个图来了解一下填料合成的整个过程:
![]()
以C18的填料合成为例,C18的氯硅烷与硅胶表面的硅羟基发生反应,C18硅烷基取代硅羟基中的氢原子在硅胶的表面键合上C18长链。完全硅羟化的硅胶表面硅羟基浓度为8µmol/m2,因为C18硅烷基的体积远比一个氢原子大得多,由于空间位阻的作用,通常与C18硅烷基发生反应的仅达2~3.5µmol/m2,也就是说只有不到50%的硅羟基被反应掉,剩下的50%多的硅羟基残留在硅胶表面。由于硅羟基容易对样品分子产生次级保留效应引起拖尾,因此需要将硅羟基反应或屏蔽掉,去掉这个作用位点以消除次级保留效应,通常的办法是用小分子的硅烷跟硅羟基反应,如图解中的三甲基氯硅烷,使硅羟基中的氢变成了三甲基硅烷的一个惰性基团,反应掉了很多硅羟基,同时未能反应的许多硅羟基也被屏蔽掉,有效阻止了样品与硅羟基接触的机会,避免了次级保留效应的产生。但不管怎么样三甲基硅烷还是比氢原子大得多,同样由于空间位阻的作用不能将所有的残余硅羟基反应掉,既然存在就仍然有跟样品分子接触的机会,相同的样品在不同品牌的柱子上产生拖尾的严重性不同,主要的也就是接触硅羟基的机会大小的问题,从填料合成的角度而言就是键合方式是否紧密、封尾是否彻底,紧密的键合和彻底的封尾能显著减少这种机会,因而改善峰形。美国Welch公司Ulimate品牌的XB-C18、AQ-C18和Xtimate C18采取的就是紧密键合和彻底的双峰尾工艺。
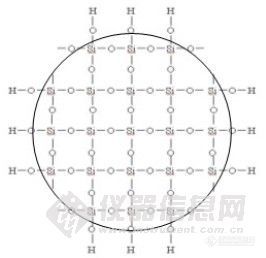
为什么残余硅羟基会引起拖尾?我们先了解一下硅羟基的结构:Si-O-H,硅胶基质上硅的四面都是氧原子,如下图:
![]()
在表面上连接有硅羟基的硅原子来说,其中一个键连接了一个羟基,其它三个键都与一个氧原子相连,由于氧的电负性比硅强,三个氧原子对硅原子产生了吸电子效应,因此对于硅上的羟基而言,这个硅原子对羟基是一个吸电子基团,使得硅羟基具有一定的酸性,其pKa约为4.5~4.7。根据电离规律,流动相的pH-pKa>2即pH>6.7时,99%以上的硅羟基应该是呈离子状态的,即Si-O- ,而pKa-pH>2即pH<2.5时,酸性环境抑制了硅羟基的电离,99%以上的硅羟基应该是呈分子状态的,即Si-OH,但其极性仍然存在,即Si-Oδ-Hδ+。
发现峰形后拖时,本人建议试试一下几种解决方案,以消除峰形后拖的状况、改善峰形。
1. 先检查一下是否是样品过载
样品过载,通常会引起峰形变差,导致前拖、后拖或平头峰,如果出现平头峰、峰高超过2000mAU或峰很高的同时又前拖、后拖得很厉害通常都认为是过载的,但这一点也并不绝对,因为不同化合物的吸收强度不一样,如果一种化合物在某一波长处有强吸收,即使出现了平头峰,样品也不一定过载,这与仪器和软件有关;相反如果吸收弱,则高浓度条件下峰也不见得有多高;所以样品是否过载应该结合浓度、进样体积和峰形一起来判断。通常认为峰高在100mAU左右比较合适,不至于因过载影响峰形。由于样品过载引起的峰形后拖不容易消除,也可能根本无法消除,继续采用以下的峰形改善措施可能不会有什么作用,而且在过载的情况下无法看清正常浓度时样品真实的分离状况和峰形,因此需要优先考虑是否过载,否则继续往下调整峰形的努力将无任何意义。
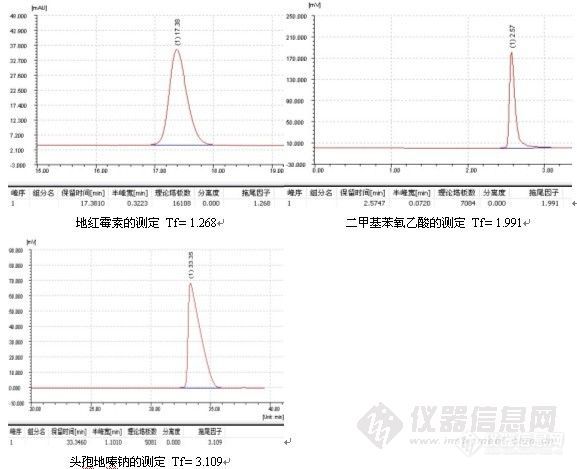
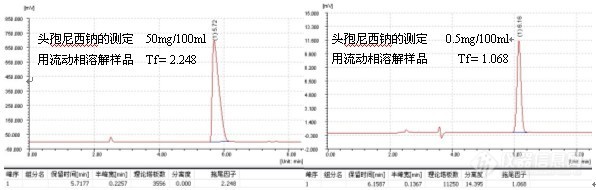
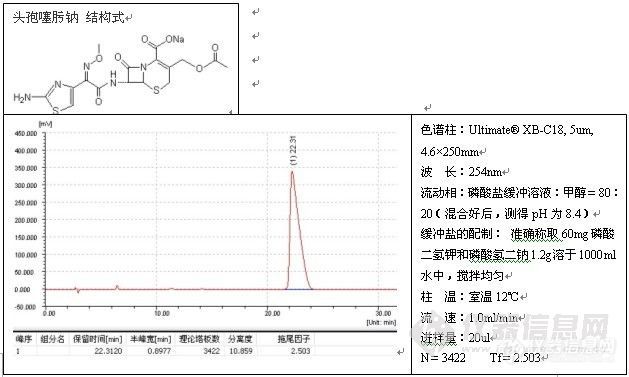
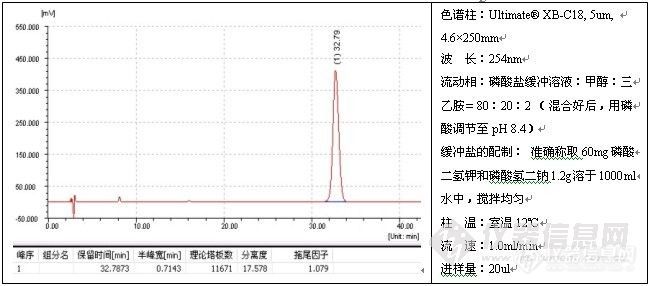
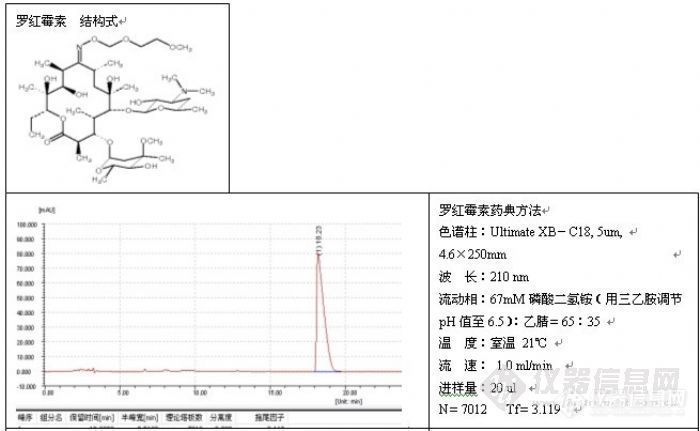
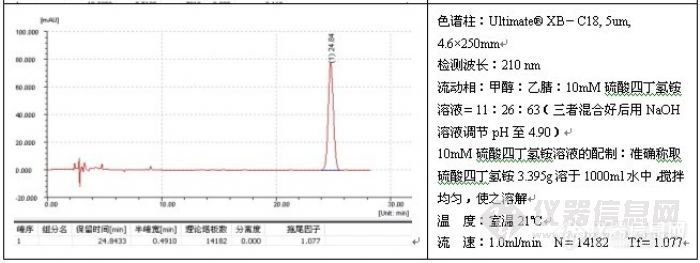
如果发现过载,需降低进样量(包括进样体积或浓度),但不管怎么样,减少进样量通常对改善峰形总是有益的,一般而言,进样量越小,峰形越好,例子如头孢尼西钠的测定:
色谱柱:Ultimate® XB-C18,5um,4.6×250mm;
波 长:272nm;
流动相:0.01mol/L磷酸二氢钾溶液(用磷酸调节pH3.5):乙腈=84:16;
温 度:室温17℃;
流 速:1.0ml/min;
进样量:20ul;
![]()
2. 往流动相中加入酸
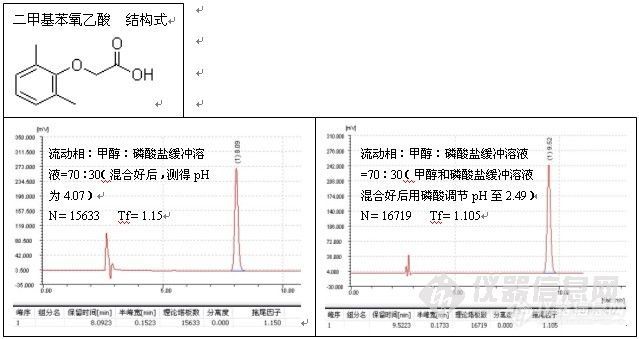
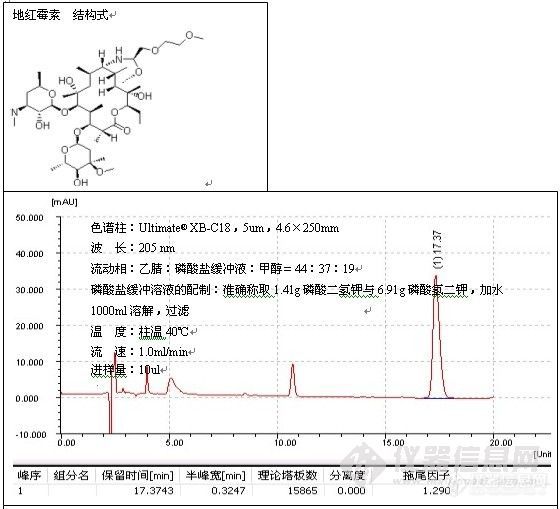
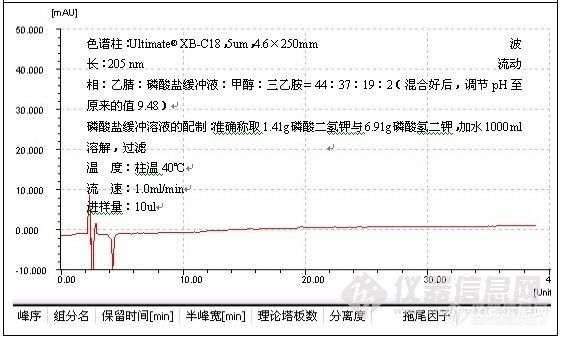
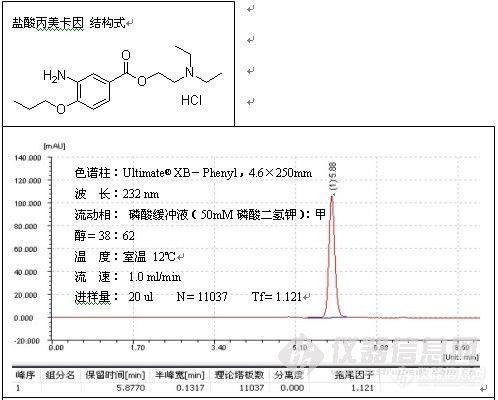
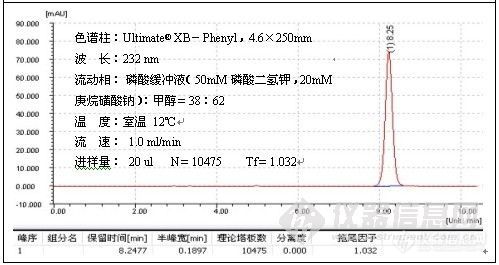
根据这一解释,往流动相中加酸将流动相的pH调至2.5以下有助于改善峰形,这种方式,主要用于改善对带羧基化合物的拖尾,例子如二甲基苯氧乙酸的测定:
色谱柱:Ultimate® XB-C18,5um,4.6×250mm;
波 长:280 nm;
磷酸盐缓冲溶液的配制:准确称取磷酸二氢钠7.8g溶于1000ml水中,搅拌均匀,使之
溶解,用磷酸将pH值调至2.50;
温 度:室温26℃;
流 速:1.0ml/min;
进样量:20ul;
![]()
由谱图可以看出,分析二甲基苯氧乙酸时流动相中加酸调节pH至2.49改善了峰形,加入酸的作用是,一方面抑制了硅羟基的离子化,另一方面也抑制了有机酸的离子化,因而削弱了-COO-与Si-Oδ-Hδ+的相互作用,减小了拖尾。