后续将有专属客服与您沟通!
关注微信公众号查看留言进度 接收留言处理通知
0
ID:houjjun
行业:其他
积分:0升级还需100积分
声望:0升级还需100声望
注册时间:0000-00-00
最后登录时间:0000-00-00
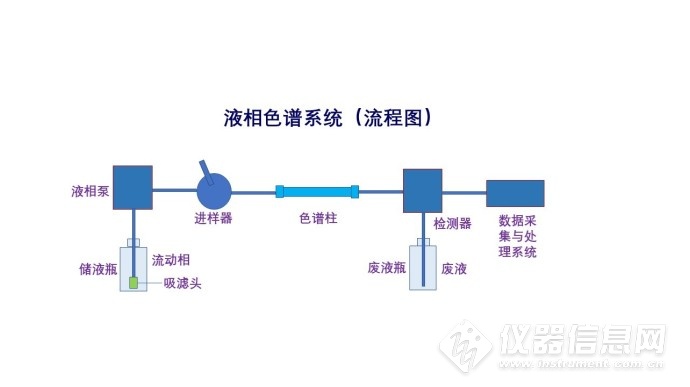
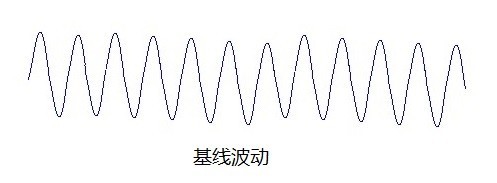
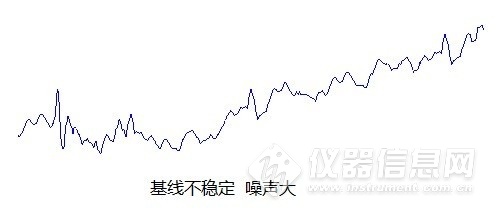
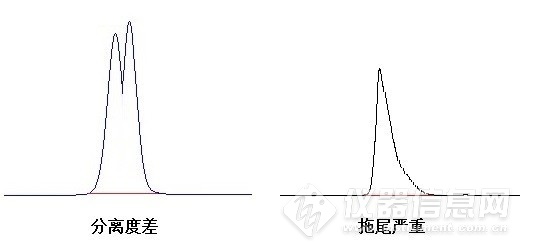
液相色谱常见故障及解决办法分享
ID:654456
ID:baby073125
原文由 石头雨(baby073125) 发表:对有该类设备的实验室很有帮助。
原文由 valorb(654456) 发表:学习下!!!!
ID:Ins_2e1e84f3
ID:yinzheng02163
ID:v2771427
ID:dadgoh
原文由 Ins_2e1e84f3(Ins_2e1e84f3) 发表:学习了,谢谢
原文由 yinzheng02163(yinzheng02163) 发表:进来学习下