X射线荧光光谱法(22)
3 定性分析方法 从X射线管产生的一次X射线照射到样品,样品中含有的各元素其吸收限的能量小于入射线能量时,都同时产生X射线荧光。
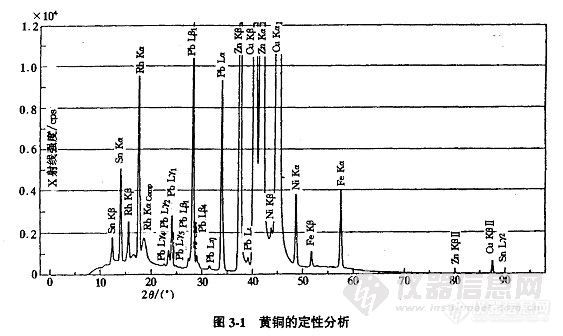
定性分析就是用测角器进行角度扫描,通过晶体对X射线荧光进行分光,用记录仪将顺次出现的谱线自动记录在谱图上,再解析谱图以获知样品中所含的元素。
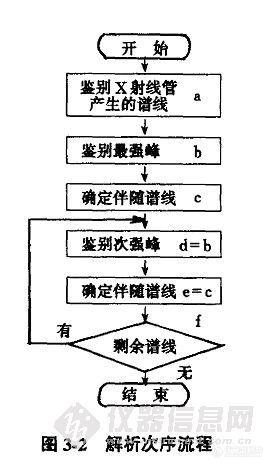
目前不少仪器都配备了自动定性分析的软件,具有自动扫描、自动解析的功能,因而可自动进行定性分析,十分方便。在这里谨对人工定性方法和步骤作一简单介绍。
1 测定条件的设定
1.1 激发条件和X射线光路的设定
作重元素的定性分析使用钨、钼、铑靶X射线管,轻元素则使用铬靶或铑靶X射线管。电压调在40~60 kV之间,当分析元素含量高时将电流(mA)调小点,进行微量元素的分析时则调得大些。还有一种电流不变,而用衰减器来衰减强度的办法。
X射线光路一般都用真空光路。但当样品有飞散危险或用大气液体样槽作液体分析时,需要在大气或氦气光路下测定。
1.2 分光条件的设定
定性分析中的狭缝系统一般都使用标准狭缝系统。分析晶体和探测器的组合及测角器的扫描角度范围示于表3-1中,这是。9F~92U等全部元素进行定性分析时的条件。
![]()
1.3 测量条件的设定
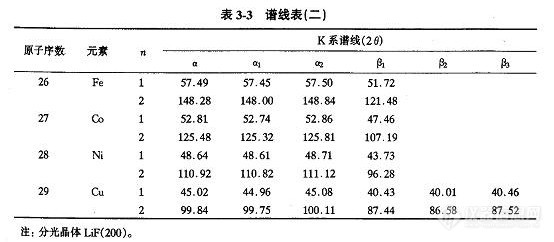
把脉冲高度分析器设定成微分测定方式。闪烁计数器的脉冲高度调整用金属铜作试样,用LiF(200)对 Cu Kα分光,增益放于适中的3档,调 SC的 HV,使平均脉冲高度值调到200/1000刻度处。流气式正比计数器系统中,用金属铝作试样,用 EDDT(或 PET)对AI Kα进行分光测定。跟闪烁计数器同样,调整PC的HV使平均脉冲高度值调在200/1000。
关于基线和道宽的设定,在闪烁计数器系统中,如果对Cu Kα线道宽设定得窄一些,那末对流气式正比计数管也可能利用同一条件。
测角器的扫描速度、记录纸速度及时间常数间有一系列的关系,表3-1列出了这些关系。用记录仪记录微量元素时,把测角器扫描速度设在 1或者 1/4(θ/mm),时间常数也增大成 1或5,缩小定性谱图的满标度,使部分放大以进行测定。
在作定性全分析中,规定满刻度的一般方法,在粉末压片样品情况下确定计数率计的范围,是使白色X射线的最大散射强度(背景)在记录仪位于50~80的刻度上。在金属样品的情况下规定满标度的原则是让主成分超出标度,分析谱线收纳在记录纸内。作轻元素的定性分析时也可采取与金属样品同样的方法(上述内容仅供参考,如果分析目的明了,根据分析目的决定)。