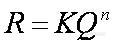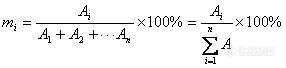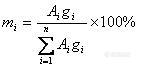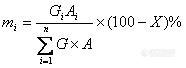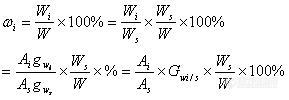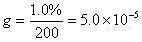板油提问的问题:
前面是问题,后面是提问的板油
1、内标法的校正因子有没有规定要求。我现在的校正因子1.9多了(qryangqiang)
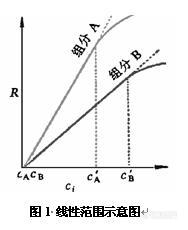
2、有时候用峰高定量和用峰面积定量会有很大差距,为什么?有时候同一个样品进样峰高和峰宽会不一致,但峰面积相差不大,为什么?检测器的线性范围是怎么确定的?分离度不好时是采用峰高定量还是峰面积定量好?归一化法中校正因子的确定文献资料查出的数值是完全可信的吗,有没有方法验证?(ionbaby)
3、在很多教材上都有氢焰和热导的质量校正因子(苯为内标),请问这些数据可以直接使用吗?(zhchen1130)
4、一个简单的例子,我们用热导测95%乙醇的乙醇含量,进样之后乙醇峰拖尾严重,严重影响计算结果的准确性,有什么办法能够计算准确点吗?(zhchen1130)
5、有时候我做的样品出峰时,在峰前会有像拖尾一样的形状,即慢慢的上升,达到峰顶,然后一下子竖直的掉下去。这是为什么呢?(3324974wang)
6、我做的内标曲线的线性达到99以上,但是在真的做样的时候总是说是无效曲线,这是为什么呀?(2000zhyy)
7、我想问的是在做甲醇残留时,因为甲醇含量很低(10ppm),我用内标发分析时,做方法验证,重现性很差是怎麽回事?(yankunsu)
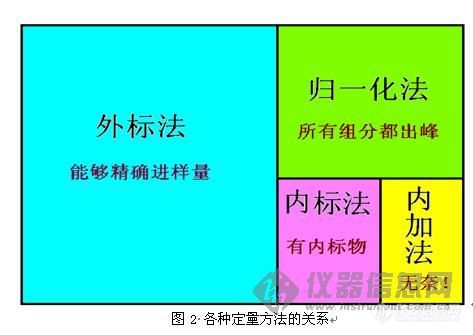
8、内标法与内加法有什么区别?内标法与外标法有什么区标?内标法和外标法两者哪个操作起来更加方便?(xuyuanjin2001)
9、请问
气相色谱能够绝对定量吗?(qingganglindeya)
10、同样的气体样品,如何保证出峰时间的一致?是进样的差异吗?(nikang3148)
11、有时由于标样难以得到或其他原因,在要求不太高时,采用利用有效碳数来计算校正因子(如酸,多元醇,含硫化合物),应注意什么?(jimzhu)
12、我现在想测定一类物质中的溶剂(二氯甲烷、四氢呋喃、乙酸乙酯等),该类物质沸点较高在
气相上不出峰,因此我决定采用外标法,没有顶空装置,我直接配成标准溶液来做,选的溶剂是出峰较晚的1-甲基-2-吡咯烷酮,我这样做需要注意的地方是什么?什么地方可能会带来较大的误差?(lpr20)
13、多点内标是怎么弄的?能赐教吗?(thysy2008)
14、现在我有一个样品,其中含有5个组分,现在我只有其中的3个标样,其他没有,我想用外表法测出这5种组分的含量,但是其他两种没有标样,无法知道其校正因子。请问我该怎么做?(aiqing1981)
15、我想检测一个混苯中的组分(含有甲苯\二甲苯\苯\及其它低沸点\高沸点组分),如何定量?(我们采用程序升温50-200度),另外我想请问
气相出峰的先后与样品沸点有必然联系吗?(chdongdong)
16、为什么“对于无法得到校正因子或者未知组分,利用它距离它较近的峰的相对校正因子即可”。有什么理论依据吗?(aiqing1981)
17、在开机升温过程中,未点火时出现检测器有信号,有一次信号在10.5,现在多时在0.2!不知道是什么原因?(gxh0601)
18、检测时出现两次检测的基线不同,且相差很大!请问原因?(gxh0601)
19、前面有人提到内标法线性相关系数要大于0.9997?是不是太高了啊。。一般不是三个九就可以了么?(helixsun)
20、水中甲胺磷用什么试剂提取效果比较好,二氯甲烷,氯仿,苯等效果都不是很好。(wscj8888)
21、我的仪器是7890.检测器是FID.我现在要测废溶剂中的丙酮含量(其含量大概有85%).请问如何配标准曲线,样品含量太大了直接进样会过载,样品应如何处理?(gzeternal)
22、您有没有做过茶叶中的有机磷农药检测,主要是乐果、敌敌畏、杀螟硫磷、喹硫磷和乙酰甲胺磷5中农药,我们单位用的是瓦里安CP-3800型号,不知道您是否能指教一二。偶最近在做这几个参数,要是结果不理想,再具体请教您(banana_hj)
23、我对一个甲醇中间控制的样品准备作分析,主成分是甲醇,含有醇、醚、酯、烃等,采用外标,由于组分太多,标定比较麻烦。所以我想了一个方案,就是先测出其他组分对某一组分的相对校正因子,然后在中控分析时每次只要标定这一个组分,可以计算出其他组分。这实际是2步计算,第一步是外标法计算出这个组分含量,第二步是用这个物质作为内标来计算其他物质。
我不知道:
1:这样的方式是否会引入较大误差?
2:用安捷伦的2070软件是否能够实现这个功能,直接出报告?(yaofei)
24、在使用
气相色谱分析时,有的物质分离不开,可以通过什么方式使其分开?(lyf0068)
25、请问相对较正因子如何定义,如何做才能更准确?(xiaojunzhang)
26、我们的NPD的出峰看上去还可以,可是积分出来的峰面积只有几到几十,这是为什么?(hongyue)
27、我的色谱为Agilent 6890N,检测器为FPD,不知为何,点火十分困难,往往是熄火后,再过一段较长的时间才能点着火,且还不十分稳定?向agilent公司咨询过数次均不见明显的效果。用过的方法有减少H2流速,提高空气流量,降低载气流速等。(davis_0411)
28、内标法与内加法有什么区别?内标法与外标法有什么区标?内标法和外标法两者哪个操作起来更加方便?什么时候使用内标,什么时候使用外标?(xuyuanjin2001)
29、
气相用多氯联苯混标做标准曲线时候,谱图的峰分离度很小,该如何调整?(xiangming)
30、我用岛津QP2010PLUS
气质测PBDEs(外标),发现标准溶液的重现性实在是太差了。
标准液是在中国计量院买来的,应该不会有问题,1ml/支,然后我用分析纯的丙酮稀释成了50ml作为标准液用,现在用的单点工作曲线,发现标准液重现性极其的差。但是上个星期我们的
气质刚做完校准,也是中国计量院的人给做的,重现性却非常好,RSD在百分之一点几。人家说我们的仪器没有问题。
那么是哪里有问题呢?请您给点帮助。感激不尽!我稀释标准液是不是不能用分析纯的稀释?分析纯的丙酮会对重现性产生影响吗?换成色谱纯有用马?(mitsumi)
31、测白酒时,在甲醇峰和乙醇峰之间有一未知物影响对甲醇的定量,很难分开,有什么好办法解决?(rxlwhite)
32、我想知道流速对于浓度型检测器面积带来怎样的影响(就比如TCD)?如果进样量一样,一定浓度的组分通过TCD时,带走的热量一样,理论上都要补偿同样的能量,面积也应该一样,是否是同浓度的组分,峰高是一样?为什么一样?(lvqiang_2001)
33、
气相色谱的检测器的单位换算关系,不知pg/sec与mg/l的关系?(plank)
34、
气相用多氯联苯混标做标准曲线时候,谱图的峰分离度很小,该如何调整?(xiangming)
35、
气相的仪器检测限应该如何定? 我们做有机磷农残时,进单个的高浓度标样都有峰,可配成混标走程序升温有些标准物就没峰了,比如我配的5个标准品混标,可进样后才出3个峰,这是为什么?(shetingting)
36、我现在用的
气相色谱是GC-Clarus 500, 像这样的仪器可以进行gage R&R(仪器的重复性和再现性)的分析评价么?
我公司一般对天然胶使用丙酮抽出,对混合胶料用甲苯抽出,我想请教一下对于抽出物如何进行GC的分析呢?(zhqf)
37、我用TCD检测器,手动进样,用内标法定量时,标准曲线的相关系数有时不好,请问是进样技术的问题吗?我该注意些什么?(xdqyxhg)
38、我准备用FID检测仪器测定空气中TVOC,因为没有气体配样器,所以需要用液体定量法,需要用溶剂配标准溶液,看到的资料都笼统的说用适合的溶剂,没有说具体的,请问用哪种溶剂合适呢?(jianpingzheng)
39、外标法的标准曲线需要通过原点吗?R2=0.991可以用来定量吗?(fenglikxy)
40、测定相对校正因子的时候,如何才能获得准确的结果?(vanvan)
41、做同试样,用内标法和外标法,两个结果有差异,应当相信那个呢?(nerd)
42、我想分析CO+H2合成低碳醇的
液相产物,主要是烃类和醇(C1-C5)的混合液体,请问RTX-1 的柱子可以分析吗?如果不行需要怎么选择,选择的依据是什么?(pink007111)
43、我用内标法做有机挥发物含量的检测,为什么进样量不同时,得出的结果差距较大?(hrt001622)
44、做标准工作曲线的时候能够采用二氯乙酸甲酯和三氯乙酸甲酯做而不进行衍生化预处理?
如果用二氯乙酸甲酯和三氯乙酸甲酯直接做标准工作曲线,这样得到的曲线线性可能会好些,但可能会引入系统偏差;而如果用二氯乙酸和三氯乙酸衍生化然后在做标准工作曲线,这样得到的曲线线性不一定好,但可以基本忽略掉衍生化带来的系统偏差
2 能否用外标法代替内标法做标准工作曲线?(wangsht4)
其他问题在2楼中