后续将有专属客服与您沟通!
关注微信公众号查看留言进度 接收留言处理通知
0
ID:v2960432
行业:其他
积分:0升级还需100积分
声望:0升级还需100声望
注册时间:0000-00-00
最后登录时间:0000-00-00
请确认联系方式
请输入您的联系方式
提交留言即视为您同意遵守 《服务协议》和 《隐私权政策》
ID:emoc98311
ID:v2989356
ID:bingwang228
ID:v2826867
ID:zhonghuashendun
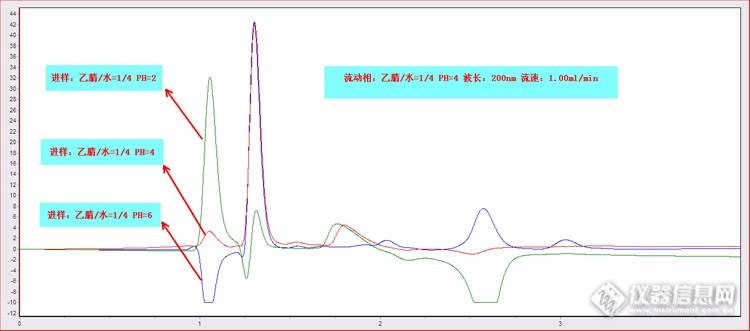
原文由 武灵(zhonghuashendun) 发表:1.05min处是否应该理解为死时间出的峰?
原文由 老多_小多(emoc98311) 发表:我认为200nm测定有很大影响,同样的色谱图,估计换个254,也许会完全不一样
原文由 orange85(v2989356) 发表:对pH,首先要注意色谱柱的pH使用范围,这是色谱柱使用基本保护;从谱图上看,pH为4时和流动相接近,所以色谱图好点,但也有好多峰啊,可能是色谱柱有关系,如果是脏了,得好好冲冲柱子。
原文由 夏天的雪(bingwang228) 发表:1,低波长检测时流动相或者样品中的杂质可能被检出,显得杂质比较多,2,ph越低1.05min时的峰越高怀疑与加入的磷酸有关,3,后面倒峰的出现可能也和磷酸含量有关,之前有过误操作,测乙酸含量时用乙酸调了ph值结果出现负峰
原文由 vm88(v2826867) 发表:出负峰与PH有关系吗?