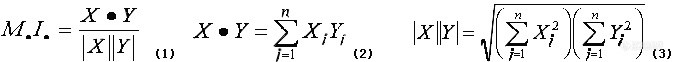三、近红外光谱分析法(Near intra-red spectrum,NIR):波长范围800~2500nm(12500~4000cm-1),优点:1、没有中红外光谱(Mid intra-red spectrum,MIR,4000~400cm-1)吸收带显示出的边缘干扰(fringe interference),故在一较大的吸收动态范围内这些吸收带强度与被测物浓度之间有线性关系;2、和MIR不同,水分吸收不会覆盖C-H、N-H和O-H的吸收带,因此,NIRS(NIR Spectroscopy)可用于水溶液样品、含水固体和泥浆状样品的分析;3、样品可不经溶剂稀释、制备溴化钾片等处理而可直接测定,免除样品制备时可能带入的误差,因而其定量效果优于GC、HPLC和FTIR等法。
药典中原料药多用MIR作指纹分析,或以标准图谱核对,或与参比标准对照,所得光谱质量多取决于样品制备的准确性和重现性,而且操作繁琐、影响因素很多。NIRS法则方便得多,仅需将样品装入样品池内而不必作预处理。一般,液体样品可直接装入光路长1~30mm的石英池中。固体样品可直接放入具有石英窗的反射样品杯中;盖上弹簧压缩盖即可。在1分钟内即可完成装样;取出样品、清洗杯子的时间也不需1分钟。通常将样品杯置入NIR光谱仪内,每秒进行5次扫描;一般扫描50次求得平均值制得最后的光谱,继之将光谱数据进行加工以作数据的定量或定性的解释。通常一次NIR分析仅需2~3分钟。
(一)用于鉴别:目前常用的是借助于计算机辅助的光谱匹配法(Spectral matching),就是将供试药物的光谱和存贮于机内的参比光谱进行对比并计算出“匹配系数(Match index,M.I.)”。若M.I.越接近1.000,则这两个光谱越是匹配;而当M.I.接近于0.000时,则为极不匹配。有时会出现负值。
![]()
式(1)中,X和Y是二个光谱的矢量表示(Vectorial representation); 式(2)中Xi是光谱A在波长i处的振幅(吸收度);Yi是光谱B在波长i处的振幅(吸收度)。矢量X和Y是由光谱A和B的N个波长处所得吸收度读数总和而得到(在某波长范围内每隔Mnm测一次吸收度,故有N个波长点)。在N维空间中这二个矢量具有不同的大小和方向。这二个矢量间可形成一个夹角(q),夹角的cosq即可确定M.I.。若q=0°,则cosq=1.000;若q为90°,则cosq=0.000,二矢量正交,完全不重叠。若q大于90°,则cosq为负值,这种情况有时会出现。