原创大赛进行得好快啊·~转眼就到9月份了,正好将暑期的实验总结下,来参加大赛了~~~~大家轻拍板砖~~~ 奖品许愿:容我再想几天哈~~~
摘 要:本实验用酚—氯仿抽提的经典方法从动物心脏中提取总DNA,电泳检测后进行分离纯化,获得总DNA样品。以D-loop序列为引物,用提取的总DNA作为模板,在适宜条件下PCR快速扩增mtDNA D-loop区,获得了大量的目的片段。
线粒体(mitoch ondria)是真核细胞核外唯一的遗传物质,哺乳动物mtDNA是约16.5kb的闭环双链分子,线粒体DNA的结构上有一个独特的D—Loop环(displacement loop region):位于tRNA—Pro和tRNA—Pile的基因之间,由少数碱基构成一个突出结构。在线粒体DNA上,D—Loop环是整个线粒体基因组序列和长度变异最大的区域,其进化速度最快,一般用于种内种群间的系统进化分析。线粒体作为真核生物胞质遗传的重要组成部分,mtDNA是由卵细胞传递给后代,被认为属于典型的母性遗传。
有关生物进化的研究一直是科学家关注的问题,现在国内外很多学者利用mtDNA D-loop区研究生物进化,采用Neighbor-joining算法,绘制生物系统进化树。哺乳动物线粒体DNA的D-loop区是其复制和转录的起始区域,是一个高度多态性和突变性的区域,其中D—loop重链RNA(DH-RNA)与复制和转录功能密切相关。目前有关mtDNA D-Loop的遗传变异分析的研究已在人、牛、猪、马、鸭、鱼类、昆虫类、两栖类等多种动物中开展。为了探讨动物的分类地位,我们以位于D - loop 两端的tRNApro和12S rRNA 基因内的部分序列设计的2 对引物通过聚合酶链式反应(Polymerase Chain Reaction , PCR) 扩增mtDNA D - loop ( P1 和P2) ,通过测定产物序列,相关比较,进一步分析其分类地位,以及mtDNA D环多态性,为以后进一步研究线粒体D—Loop区的多态性与疾病的关系以及分析物种之间的进化规律奠定了基础。
1 材料与方法
1.1.1实验试剂
提取DNA:生理盐水(去除组织中的淤血)、 TES液(抑制DNA酶作用)、SDS(表面活性剂,抑制DNA酶)、饱和酚(将蛋白质变性)、酚∶氯仿∶异戊醇(体积比为25∶24∶1)、氯仿∶异戊醇(除去酚)、无水乙醇(预冷,沉淀DNA)、预冷的乙醇(洗涤DNA)、 TE(降解DNA)、蛋白酶K(降解蛋白质)
电泳: TBE缓冲液(pH8.0)、TBE缓冲液、琼脂糖(凝胶介质)、溴化乙啶(EB,荧光染料)、溴酚蓝(指示剂)、标准λDNA(48kb,Mark)、上样液(蔗糖+溴酚蓝)
PCR扩增:引物P1:5’-TATGTACCATGAGGACAAATATC-3’,P2:5’-ATTACACCTCCTAATTTATTAGGAATC-3’ [6]、dNTP(2.5mmol·L-1)、10×Buffer溶液、TaqDNA聚合酶(2.5U·μl-1)、超纯水、Marker(DL2000)、PCR产物回收试剂盒等。
1.1.2仪器
微量
移液器、Eppendorf离心机、微波炉、DYCp31型电泳仪、凝胶成像系统、恒温水浴锅、PCR仪
1.2 实验方法
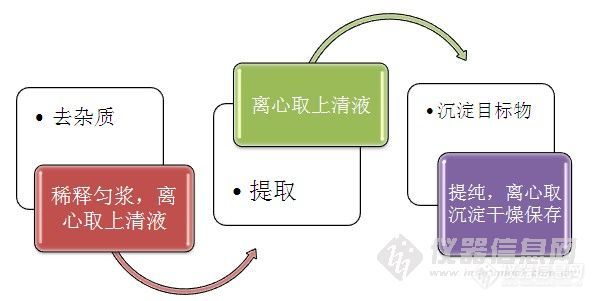
1.2.1 总DNA的提取(总的说来采取的步骤图示为:)
![]()
1.2.1.1 将材料解块,用NaCl洗去血污。剪一小块组织,剪碎,放入-20℃预冷的研钵中加入TES充分研磨。
1.2.1.2吸取匀浆液到离心管中,再加入SDS及蛋白酶K(20mg/ml)充分混匀后于56℃保温,每2小时摇一次。放置室温,加入等体积的饱和酚,颠倒混匀呈乳浊液,10000rpm离心10min(液体分层,上层为黄色,下层无色)。分离水相和有机相小心吸取上层含核酸的水相到一个新的离心管中。
1.2.1.3加入等体积酚∶氯仿∶异戊醇(25∶24∶1)380μl,颠倒混匀15min,10000rpm离心10min,取上清液移到一个新的离心管中。再加入等体积氯仿∶异戊醇(24∶1)颠倒混匀15 min。这时液体分层,10000rpm离心10min,取上清液移到一个新的离心管中。
1.2.1.4加入2.5倍体积-20℃的预冷无水乙醇沉淀DNA。于12000rpm离心15min,用乙醇快速洗涤沉淀,离心,弃上清,干燥。加入TE于40C冰箱中保存备用。
1.2.2 电泳检测总DNA
1.2.2.1称取0.24g琼脂糖于三角瓶中加入30ml0.5×TBE缓冲液配成0.8%的胶;用微波炉加热,煮沸,振荡,反复加热振摇2~3次,使糖充分融化。用医用胶布将胶床两端粘好,注意不要超过底部以免放置不平。
1.2.2.2等胶冷却至约60℃时,将融化的琼脂糖小心地倒入已插好梳子的胶床中,再使其自然冷却,直至完全凝固。小心向上方拔出梳子,去掉制胶槽两边的胶带,小心地将胶和胶床放入电泳槽。向电泳槽中加入TBE缓冲液,液面高于胶面1~2mm,以使其全部处于电场内。
1.2.2.3取8μlλDNA,加2μl上样液,混匀后,小心点入上样孔中。取4μl样品DNA,加2μl上样液在parafilm上混匀,同样点入上样孔中。将电压调至100V,接通电源,打开开关,开始电泳。大约20分钟后,待指示剂迁移到距上样孔1.5cm以外时终止电泳,切断电源,取出凝胶,放入含EB的染色液中染色20分钟。凝胶取出后,于凝胶成像系统中观察分析。
1.2.3总DNA的纯化
提取的总DNA加TE补到50μl,加0.5μl的RnaseA,37℃温浴1h;加50μl氯仿:异戊醇,颠倒均匀,大约10次;10000rPm,离心10分钟,取上清夜;加150μl无水乙醇,轻微晃动;12000rPm离心10分钟,弃乙醇;加300μl70%乙醇洗涤DNA,弃乙醇控干,55℃干燥DNA;加25μlTE溶解DNA
1.2.4 PCR扩增线粒体DNAD-LOOP基因
1.2.4.1 引物的设计
扩增引物为昆虫通用引物,引物设计参考Simon[21]等,引物由上海生工生物工程有限公司合成。
1.2.4.2 扩增体系的建立
每一样品的扩增体积均为25μl。将下列试剂按表1的顺序加入0.2mL的EP管中。
![]()
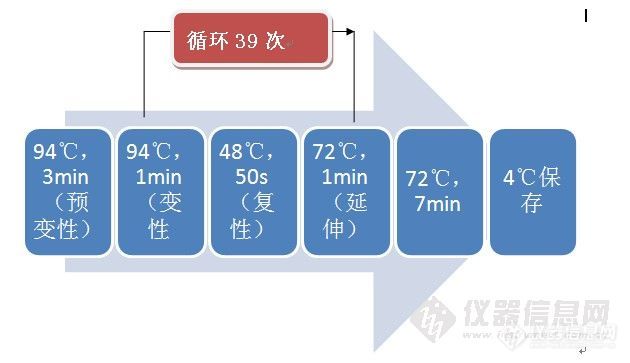
1.2.4.3 PCR扩增
对上述加好试剂的0.2mL的Ep管短暂离心,灭气泡后放入PCR仪。扩增共运行35个循环,每一循环包括:
![]()
扩增产物用1.0%的琼脂糖凝胶电泳检测其大小、纯度及浓度。
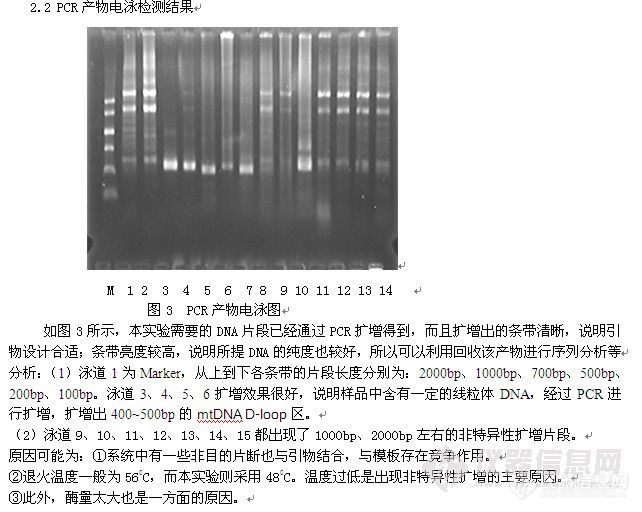
1.2.4 PCR扩增产物的电泳检测
称取0.3g琼脂糖于三角瓶中加入30ml的1×TBE缓冲液配成1.0%的凝胶。
从PCR仪中取出已经扩增的样品.用微量取液器取3μl溴酚蓝加入EP管,反复抽吸以混匀,再从中吸取3μl,加入上样孔.吸取6ulMarker点样.插好电极,接通电泳仪电源,调节电压120V,电泳20min,以指示剂迁移位置判断.将凝胶取出置于EB液中染色20min.
凝胶取出后在凝胶成像系统中拍照观察,从中检测其大小、纯度和浓度。
1.2.5 PCR产物的回收、纯化与测序
1.2.5.1 PCR产物的回收
称取0.3g琼脂糖于三角瓶中加入30ml的1×TBE缓冲液配成1.0%的胶。用微量取液器取3μl溴酚蓝加入Ep管,反复抽吸以混匀,再从中吸取25μl,加入上样孔。另外,吸取6μlMark点样。然后插好电极,接通电泳仪电源,调节电压90V或更低,电泳时间1h,将凝胶取出,在紫外灯下观察结果。
在紫外灯下割下目的胶条,按比例加入S1液300μl,50℃水浴10min,使胶块融化,每隔2 min摇匀一次。加100μl的异丙醇,混匀,50℃水浴5 min,混匀之后短暂离心。将溶好的Agarose胶移入吸附柱,12000rpm离心30S,倒掉收集管中的液体。将吸附柱放入同一管中,然后加入W1500μl,12000rpm离心15 S,倒去管中液体,再加入W1500μl后静置1min,离心15S,倒去液体离心1min。
1.2.5.2 纯化
将吸附柱放入一个干净的1.5mlEp管中,加20μl无菌水,37℃保温5 min溶解。 再于12000rpm离心,DNA溶于液体中。
1.2.5.3 PCR回收产物的检测
使用1.0%的胶,用微量取液器取2μl溴酚蓝加入2μlDNA溶液,加入上样孔。另外,吸取4μlMark点样。然后插好电极,接通电泳仪电源,调节电压90V或更低,电泳时间1.5h,将凝胶取出,在紫外灯下观察结果。
2 结果
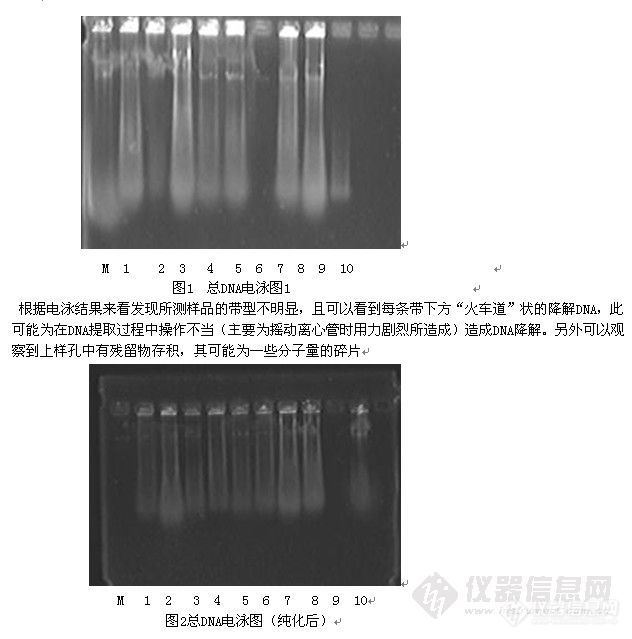
2.1总DNA电泳检测结果
结果如图1所示:提取到的总DNA跑出的条带亮度较λDNA小,与λDNA几乎在同一条线上,由此可知提取到的总DNA分子量与λDNA相似,但其含量略低于λDNA(λDNA浓度为25ug/ml)。
![]()
![]()
2.3PCR回收
2.4mtDNA D-loop区序列分析
DNA的碱基序列决定着基因的特性,DNA序列分析 (测序,sequencing)是分子生物学重要的基本技术。无论从基因库中筛选的癌基因或经PCR法扩增的基因,最终均需进行核酸序列分析,可藉以了解基因的精细结构,获得其限制性内切酶图谱,分析基因的突变及对功能的影响,帮助人工俣成基因、设计引物,以及研究肿瘤的分子发病机制等。测序是在高分辨率变性聚丙烯酰胺凝胶电泳技术的基础上建立起来的。目前最常用的方法有Maxam-Gilbert的化学降解少和Sanger的双脱氧法等,近年来已有DNA序列自动测定仪问世。化学降解法是在DNA的片段的5`端标记核素,然后用专一性化学试剂将DNA特异地降解,在电泳和自显影后,可得到从标记端延伸的片段供测读序列和进行比较。一般能读出200-250个核苷酸序列。双脱氧法是采用核苷酸链终止剂,如:2`,3`-双脱氧核苷三磷酸ddNTP(如ddTTP、ddTTP、ddGTP和ddCTP中的一种)掺入到DNA链中以终止链的延长,与掺入4种正常的dNTP的混合物分成四组进行反应,这样可得到一组结尾长衙不一、不同专一性核苷酸链终止剂结尾的DNA片段,经凝胶电泳分离和放射自显影,可读出合成的DNA核苷酸序列,根据碱基互补原则,可推算出模板DNA分子的序列。化学降解法只需一化学试剂,重复性好,容易掌握;而双脱氧法需单链模板、特异的寡核苷酸引物及高质量的DNA聚合酶,便随着M13噬菌体载体的发明和运用,合成的引物容易获得,测序技术不断改进,故此法已被广泛应用。基脱氧法的自动激光荧光测序仪,使测工作更快速和简便,而且保证高度重复性。至于RNA测序现大多采用将mRNA逆转录成cDNA后同测序,然后反推RNA序列
2.5利用DNA D-loop区序列分析物种亲缘关系
3、讨论
3.1在总DNA的提取试验中,EDTA溶剂的作用为抑制DNA活性,SDS溶液可以破坏细胞核、核膜,并分离组蛋白,除此之外还可以与EDTA共同抑制DNA活性。在饱和酚试剂制备中要在171℃条件下蒸馏,并要制为饱和状态,这是由于酚可溶于水而会对DNA纯度造成影响。苯酚可以破坏DNA的磷酸二酯键,而且可促进DNA的交联作用,10mM的TE可溶解DNA、蛋白酶K,且可溶解蛋白质除去杂蛋白。
在实验过程中组织DNA的提取中要防止用力过猛,否则会产生的对DNA的机械力剪切作用,影响DNA样品的黏度和性能。酚—氯仿抽提得到水相和有机相,核酸溶于水相中,因此在吸取水相时注意控制微量
移液器的量程,防止吸入有机相重新混入杂蛋白。在用75%乙醇洗涤沉淀时一定要快,防止样品溶于水中影响产率。
3.2组织DNA的电泳与检测中:制备凝胶时要注意倒胶虚度要快,防止产生气泡,影响样品的电泳速率。上样前要将上样液和样品DNA充分混匀,并且避免电泳后的带型不均一、整齐;点样时控制好微量
移液器的量程和垂直状态,以免破坏点样孔。电泳时随时注意观察溴酚蓝的位置,以便随时终止电泳。电泳后于EB中染色时要注意安全,因为EB是一种嵌入剂,有致癌作用。
3.3总DNA的纯化:本实验主要为前次DNA提取的后续实验,对DNA样品进行进一步的纯化以满足以后实验的需要。实验中在向溶液中加入TE酶液时,要根据个人的实际情况而定,本实验中加入15ul,在混匀时由于量少所以采用轻弹的方式振荡混匀,另外水浴时间要充分,使RNA能够得到降解,在加入无水乙醇轻微晃动后可观察到出现丝状沉淀,此即为DNA,再次离心后管壁上留有少量透明丝状物,颜色发白,即为提纯后的DNA,干燥后加入TE保存。