维权声明:本文为yuxiaofeng86原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
手上正在探究几个中间体的分析方法,现将其中的一个或几个问题,跟大家分享下,希望对大家有所帮助(谱图稍后传上)
样品:中间体(合成提纯后样品,没进行定量分析,理论认为纯度较高)
分析方法一、
流动相:乙腈+水=60+40 流速:1.0ml/min 波长:280nm 恒流 岛津LC-10AD 无在线
脱气装置流动相超声,脱气,柱平衡后进样,压力72Kgf/cm2
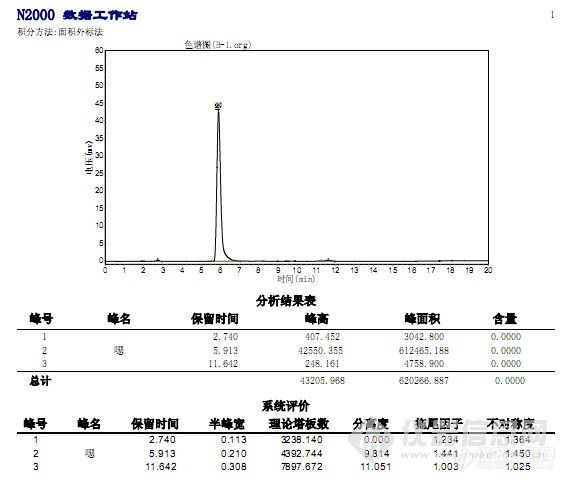
见谱图(乙腈)
分析方法二、
流动相:甲醇+水=70+30 流速:1.0ml/min 波长:280nm 恒流 岛津LC-10AD 无在线
脱气装置流动相超声,脱气,柱平衡后进样,压力115Kgf/cm2
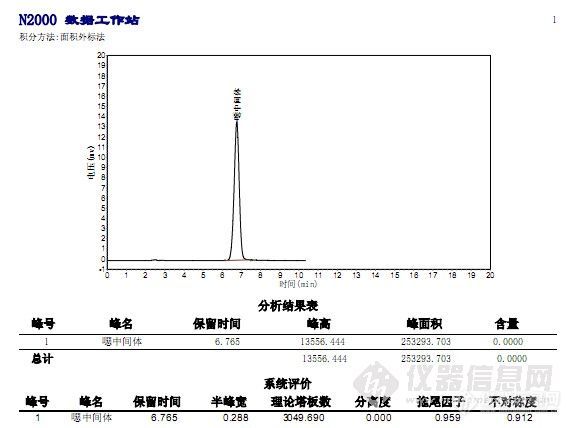
见谱图(甲醇)
根据谱图情况分析方法的优越性,极其考虑过程:
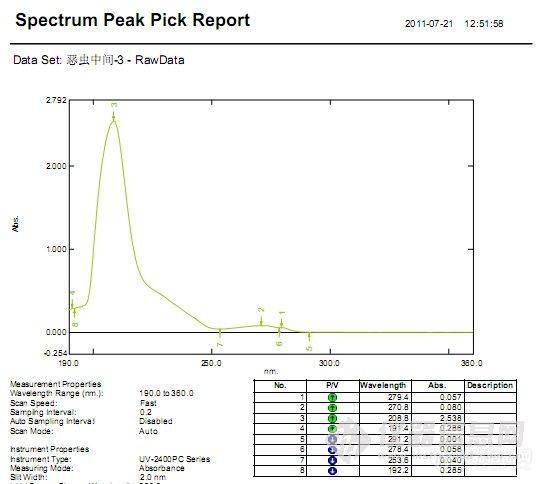
拿到样品,先了解样品的一些理化性质,稳定性,然后做个UV确定下最大吸收波长(因样品不是太纯,测的有两个)
备注:大家注意下
最大吸收波长和合适吸收波长这两个概念,合适吸收波长一般在最大吸收波长上下20nm左右,一般我们用的波长都是合适吸收波长,根据谱图进行调节;一般针对原药和含量超低的选用最大吸收波长。
确定波长后,就要选择流动相和色谱柱。80%的化合物都可以用C18 这里我就不多说了
流动相,首先选择乙腈,因为乙腈的洗脱能力强,出峰较快,能较块的知道效果,然后在进行实际情况微调。
我选择乙腈和水做流动相,进行微调后,发现分离效果不错,但是峰形却相当不好看,中等程度的拖尾。见谱图(乙腈)
通过加磷酸,不断调试PH,但对峰形没有多大影响。
后改用甲醇水做流动相,出峰效果非常好,见谱图(甲醇)
至于用甲醇代替乙腈,为什么能取得如此的效果,我是这样理解的,因为为了达到同样的保留时间,不更改其他条件的情况下,提高有机相的浓度,有利于峰形改善。
现在确定分析方法二为本产品的分析方法。
补充:UV测得两个波长,一个是270nm,一个280nm,用270nm定为检测波长时,发现主峰钱有个峰,峰高较高,影响美感,而用280nm则不会有此情况,所以确定为280nm。见图(E-3)
![]()
![]()
![]()