
获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
而异辛酸钠就是成了钠盐,性状由油状液体变成固体(易吸水),溶解度跟酸也差不多。
一开始看到溶剂沸点228℃时,GC是有点拒绝的,一个溶残飚到两百多度,你考虑过其他溶残的感受么,于是就没有太多考虑气相,而且结构里拖着一条长链(事后发现这是条拉我下坑的链!),虽然是个酸(盐),但保留应该不弱,于是毫不犹豫的设计实验方案了。
流动相哗哗哗的倒着,容量瓶发出叮叮叮的碰撞声,就像对天平打印机发起挑战,12月27日开始了第一次验证实验。称量、溶解、稀释、定容,一切都顺利的进行着,直到配制加标溶液,出现如下一幕,往供试品溶液加入异辛酸对照溶液时产生黄色沉淀。

面对突如其来的情况,我承认当时我就懵B了,更别说马上找出沉淀的原因和解决方法,试过把溶液过滤掉,但也是然并卵,放置一会又会浑浊,这样的溶液拿去进样,进样针稳稳的堵,最后只好停止配样。经过一个晚上思考,终于记起了样品的性质4(参见上文),然后就想着不产生沉淀的方法,既然遇酸就沉淀,那我就用异辛酸钠做对照吧。
流动相哗哗哗的又倒着,容量瓶又发出叮叮叮的碰撞声,12月28日开始了第二次验证实验。与第一次开始时一样,一切都顺利的进行着,当要配制加标溶液时,抖着小手往样品加入对照溶液,心率直接上120,相信薛定谔第一次遇到猫也差不多这心情,最终得到了澄清的供试品加标溶液。然后就顺理成章的把溶液配完,放进样品盘,序列运行,回家睡觉。
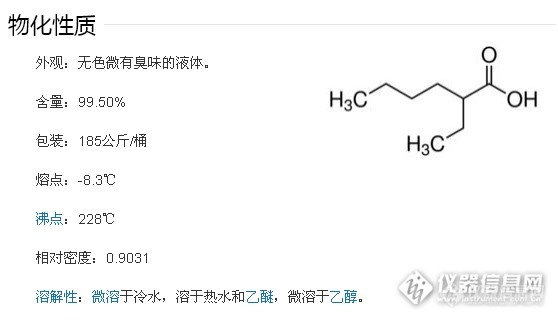
第二天上班,像往常一样打开结果看,嗯,进展还顺利,灵敏度溶液S/N有28,6针对照RSD是2.3%,空白没干扰,然鹅当我打开加标时,结果是这样的……

如上图,在异辛酸钠出峰处,加标溶液的峰明显小于对照溶液,而且在前面8min左右出一个胖胖的峰,而且加标溶液中红框的两个峰面积加起来与对照溶液异辛酸钠峰面积很接近。我再一次懵B了。

接连两次失利让我渐渐有一种液相不适合测这个的感觉,加上仪器不断高浓度进样(供试品浓度30mg/ml),造成针座出现严重残留。
由于个人原因,必须要在1月4日之前搞定这个实验。经过元旦一天的休息后,于是我开始尝试用气相,首先选一根合适的柱子,结果如下。
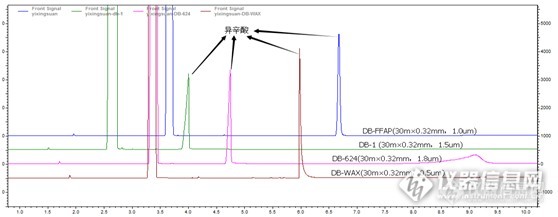
DB-1(30m×0.32mm,1.5μm)和DB-624(30m×0.32mm,1.8μm)色谱柱上异辛酸色谱峰峰形前沿;在DB-WAX(30m×0.32mm,0.5μm)色谱柱上异辛酸峰形拖尾;而在DB-FFAP(30m×0.32mm,1.0μm)色谱柱上异辛酸峰形较好,就你了FFAP。
虽然时间上很紧急,但这次我并没有急着设计实验方案和配样,先把各种溶液试水一下。既然用气相,那对照溶液还是得用异辛酸配制,so还是回到最开始的怎么解决沉淀的问题。在一次偶然的冥想中,突然记起样品性质5,于是立马动起手来,往稀释剂中混入一点甲醇,沉淀如愿消失了!
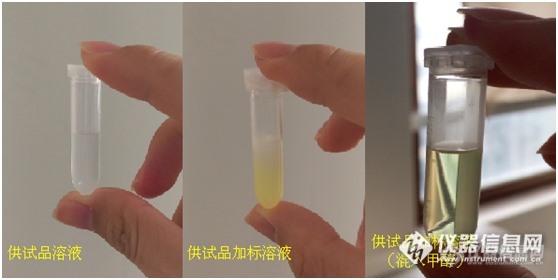
正当我认为问题解决了的时候,新的问题又双叒叕出现了!
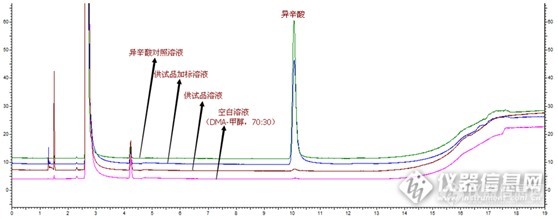
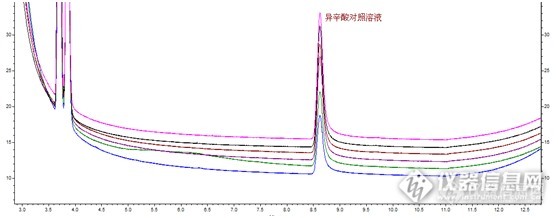
加标溶液和对照溶液这对磨死我的小妖精,前者总是比后者小一截;而且多次进对照后空白很容易出现残留干扰;连续6针对照溶液异辛酸的峰面积一针比一针大,第6针是第1针的2.5倍。
6针翻2.5倍,比股票涨停还要猛啊有木有,简直惊呆了作为实验狗的我,毕竟干了7年我的工资都没涨这么快过……
对于连番爆出的问题,我表示已经麻木了,心里毫无波澜,甚至还有点上瘾。

对于残留的问题,原因也不难分析,针对气相直接进样,残留基本上都是在衬管引起的,由于衬管表面玻璃材质以及玻璃棉灭活不完全,或者玻璃棉丝断裂产生内部横截面,不能保证整根衬管内部都100%惰性,很容易对一些性质较活泼的化合物引起吸附,而异辛酸有羧基,带酸性,正对衬管的胃口,这也是气相做酸碱性较强的化合物时,峰形往往不完美的原因之一。
原因分析了,问题解决也不难了,我决定用一个比较粗暴的方法,就是在不影响供试品的前提下,向溶液中加入一种酸性更强的酸(比如盐酸),原理就是与异辛酸竞争陈管内的吸附点,把异辛酸顶出来,从而减少对其的吸附。后来也在百度和中国药典里看到类似的2-乙基己酸(即异辛酸)的溶残方法,所以有时候遇到问题,先在网上查一下,或者翻翻药典也是很有用的,当然这是后话了。
然后就是开始尝试往配制的溶液中加入酸溶液了,所得结果如下


吸附和加标溶液的问题都消失了,终……终于可以开始设计方案做验证了。流动相哗哗…啊不对,气相不用倒流动相,1月3日开始了第3次验证实验,这一次顺利完成了!最终方法如下:
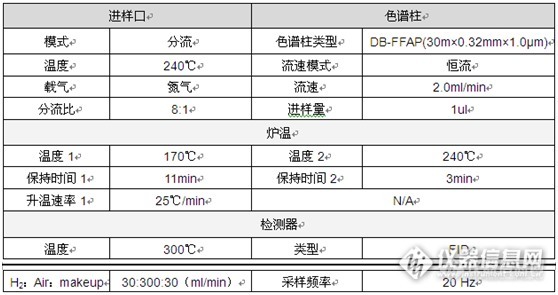
这是最终溶液配制方案:

最终的验证结果(更具体的峰面积和称样数据就不列出了)
1.系统适用性
空白溶液对异辛酸的测定无背景干扰;灵敏度溶液中异辛酸的信噪比为47.0(≥10);对照溶液连续6针的异辛酸峰面积的RSD为2.52%(≤10.0%)。系统适用性符合检测要求。
2.专属性
空白溶液与供试品溶液及100%供试品加标溶液比较,空白溶液在对照溶液和供试品溶液的异辛酸峰保留时间处无干扰;供试品溶液及供试品加标溶液与对照溶液中异辛酸溶剂峰的保留时间一致;与供试品溶液相比,供试品加标溶液色谱图中异辛酸保留时间处峰面积明显增强。方法专属性良好。
3.精密度
(1)分析重复性
由分析人员甲配制的6份分析重复性试验溶液中,异辛酸含量的RSD值为2.43%(≤10.0%)。
(2)中间精密度
由分析人员乙配制的6份分析重复性试验溶液中,异辛酸含量的RSD值为2.06%、(≤10.0%);分析人员甲与乙分别配制的12份分析重复性试验溶液中,异辛酸含量的RSD为2.15%(≤10.0%)。方法精密度良好。
4. 定量限和检测限
异辛酸定量限溶液浓度水平为250ppm(≤500ppm);该浓度水平异辛酸溶剂峰的信噪比大于10,连续3次进样峰面积的RSD值小于10.0%。方法定量限满足检测要求。
异辛酸检测限浓度水平为100ppm;该浓度水平异辛酸溶剂峰的信噪比大于3。
5. 线性及范围
异辛酸从定量限至限度水平的200%呈线性关系,线性相关系数r分别为0.9998(≥0.990),Y轴截距与100%限度浓度峰面积比值的绝对值分别为1.8%(≤10.0%)。线性及范围符合异辛酸定量检测的要求。
6. 准确度
50%,100%和150%三个加标浓度水平共9份回收率试验溶液中,异辛酸的回收率单值均在80.0%~120.0%范围内,回收率单值的RSD不超过10.0%;平均回收率为98.1%。方法准确度良好。
7. 耐用性
在变化的各色谱条件下,灵敏度溶液的信噪比均大于10,对照溶液3次测定异辛酸峰峰面积RSD均小于10.0%。
实验结果表明初始柱温在168℃~172℃内变化,流速在1.8ml/min~2.2ml/min范围内变化,同一规格型号的不同色谱柱,方法耐受性良好。
好了,实验和文章到此都终于结束,对于上文,我总结一下实验过程的几点感想:
(1)对于研发人员来说,样品性质都是未知的,要善于发现和联想,说不定几个月前出现的一个小现象就是解决紧急问题的关键;
(2)由于时间相当紧迫,实验中很多细节未进行进一步研究和优化;
(3)第1次验证中,假如在稀释液中也混入甲醇,沉淀问题就能解决,只是当时完全没想起样品的性质5;
(4)第2次验证中,发现加标溶液中异辛酸前面出一个胖胖的峰,其实可以接出来送MS,进而分析原因;
(5)最终的方法中,甲醇的比例,加入盐酸的比例以及盐酸溶液的浓度都未摸索过(时间紧迫);
(6)最终的方法中,异辛酸峰形还是有点拖尾,如果把色谱柱DB-FFAP(30m×0.32mm,1.0μm)换成0.5μm膜厚,也许能得到峰形更好,耗时更短的方法(时间紧迫,拿起一根觉得可以就用);
(7)对于新药研发分析人员,查阅文献的能力固然重要,但偶尔翻翻药典的正文,也许会有新的发现,如果我在之前看到药典的方法,也许就能少一半工作量,以及码少一半字;
(8)第一次发原创,重在参与,写的好的,能给大家提供点思路或者什么的固然是好,写的不好的,也烦请尽管提出来;
(9)最后的最后,吐槽一下仪器论坛这个话题发表编辑界面,虽然这里主要是发科技文章,但和微信公众平台的编辑界面比较,真是难看兼不好用












































 。。。
。。。
