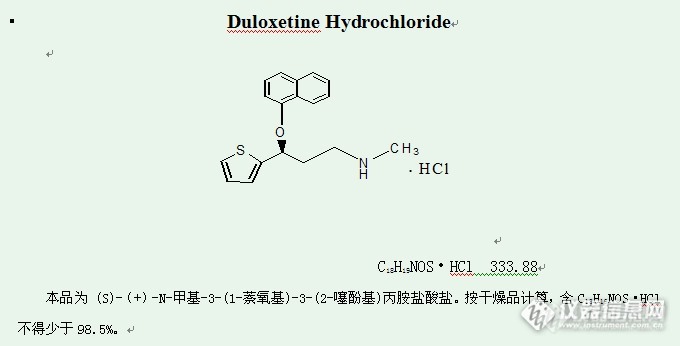
百花齐放斗艳,Welchrom® C18与ZORBAX对比
目前,国家审评中心对申报品种的有关物质要求更严格,现在好多人都挂了,有些是挂了彩,有些是全挂了。。。 国庆期间不说这个,权当瞎扯。
当前形势逼人,有关物质一定要认真研究,特别是和标准图谱对比,原研对比以及标准对比。下面以实例分享,标题就不列出药物名称啦。
色谱柱信息:welchrom c18 pn:00310-02041,sn:w132111841 4.6*150mm实例分享:
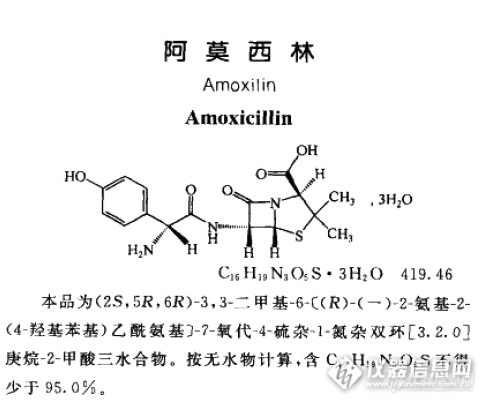
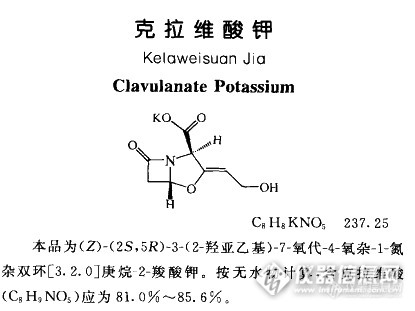
1.阿莫西林克拉维酸钾
![]()
![]()
色谱条件:
有关物质 取本品的内容物适量,精密称定,加流动相A溶解(必要时冰浴超声助溶)并稀释制成每1ml中含阿莫西林2mg的溶液,滤过,取续滤液作为供试品溶液;精密量取适量,用流动相A定量稀释制成每1ml中含阿莫西林40μg的溶液,作为对照溶液。照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定,用十八烷基硅烷键合硅胶(A型)为填充剂;流动相A为0.01mol/L磷酸二氢钾溶液(用2mol/L氢氧化钠溶液调节pH值至6.0),流动相B为0.01mol/L磷酸二氢钾溶液(用2mol/L氢氧化钠溶液调节pH值至6.0)-乙腈(20:80),流速为每分钟1ml;检测波长为230nm;先以流动相A-流动相B(98:2)等度洗脱,待阿莫西林峰洗脱完毕后立即按下表进行线性梯度洗脱,tR为阿莫西林主峰的保留时间,约为10分钟。取阿莫西林克拉维酸系统适用性试验对照品,加流动相A溶解并稀释制成每1ml中约含2.5mg的溶液,取20μl注入液相色谱仪,记录的色谱图应与标准图谱一致。取对照溶液20μl注入液相色谱仪,调节检测灵敏度,使阿莫西林峰的峰高约为满量程的25%;再精密量取供试品溶液与对照溶液各20μl,分别注入液相色谱仪,记录色谱图。供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液两个主峰面积和的1.25倍(2.5%),各杂质峰面积的和不得大于对照溶液两个主峰面积和的3.5倍(7.0%),供试品溶液色谱图中任何小于对照溶液两个主峰面积和0.05倍的峰可忽略不计。 时间(分钟) | 流动相A(%) | 流动相B(%) |
tR + 0 | 98 | 2 |
tR + 20 | 70 | 30 |
tR + 22 | 98 | 2 |
tR + 32 | 98 | 2 |
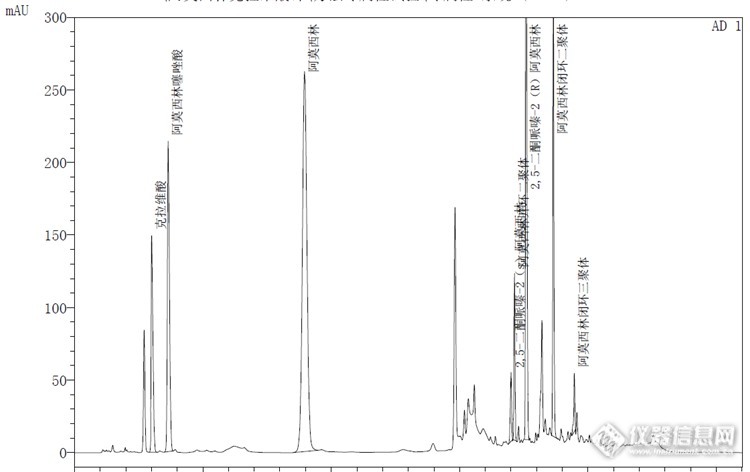
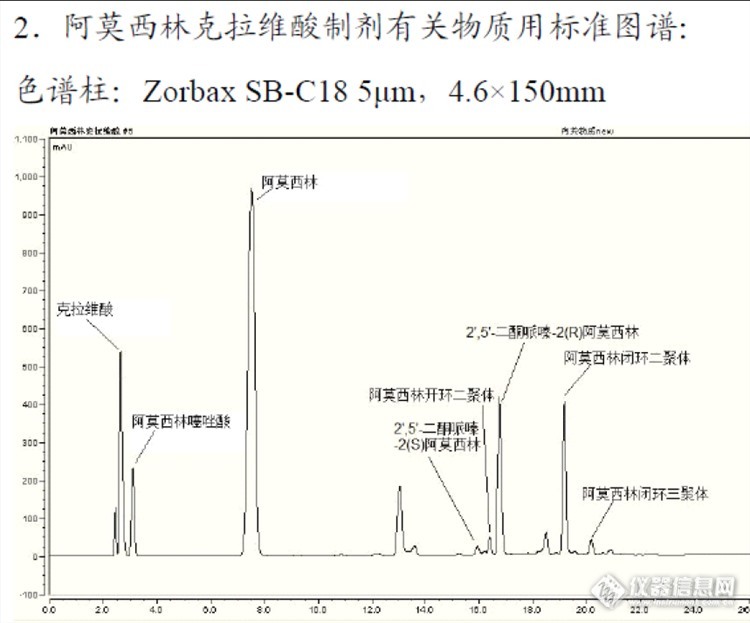
图谱对比:Welchrom® C18![]()
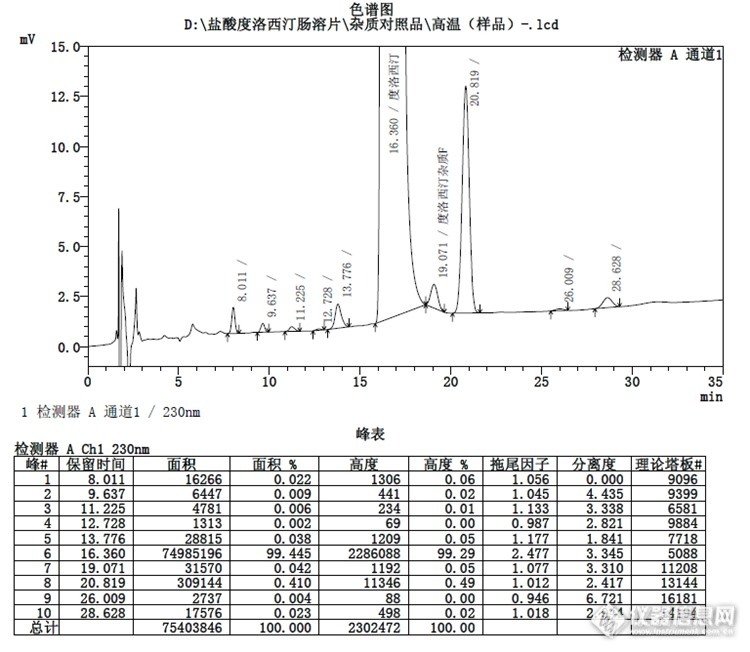
安捷伦:![]() 结论:上图可以看出月旭色谱柱峰形较为尖锐,相对较好。实例2.盐酸度洛西汀有关物质:
结论:上图可以看出月旭色谱柱峰形较为尖锐,相对较好。实例2.盐酸度洛西汀有关物质:![]()
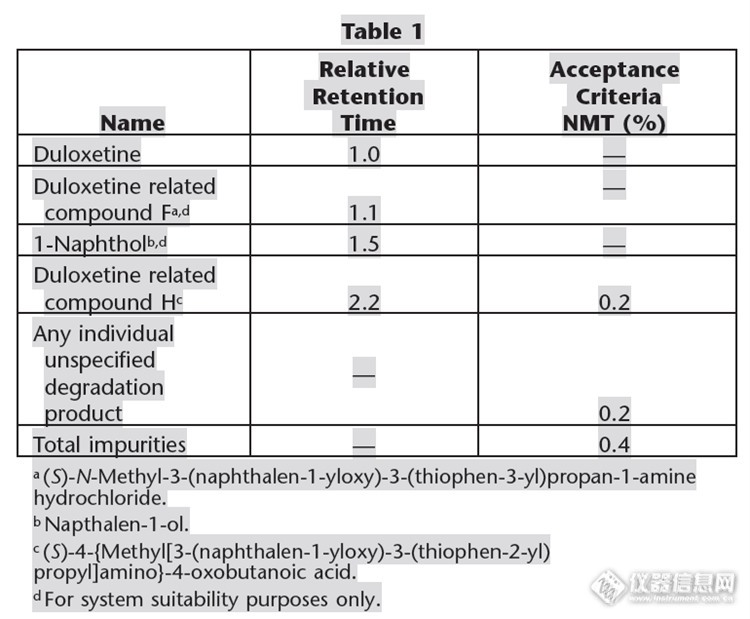
色谱条件:采用美国药典的方法,初步翻译了下确定色谱条件如下:取本品适量(约相当于度洛西汀45mg),置100ml量瓶中,加溶剂[缓冲液B(称取0.2g磷酸二氢铵和4.5g磷酸二氢钾,置1000ml量瓶中,加水适量使溶解并稀释至刻度,用磷酸调节pH值至8.0)-甲醇(50::5)]适量使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;精密量取供试品溶液1ml,置100ml量瓶中,加溶剂稀释至刻度,摇匀,作为对照溶液;取供试液水浴加热1h,作为分离度试验溶液;另取度洛西汀杂质F和度洛西汀杂质H适量,加溶剂适量使溶解并稀释制成0.5μg/ml的溶液,作为系统适用性试验溶液。照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定,用十八烷基硅烷键合硅胶为填充剂;流动相为甲醇-四氢呋喃-缓冲液A(称取3.4g磷酸二氢钾加水适量使溶解并稀释至刻度,加入15ml三乙胺,用磷酸调节pH值至5.5)(32.3:9:58.7);流速1.0ml/min;柱温45℃,检测波长230nm。取对照溶液、分离度试验溶液和系统适用性试验溶液各20μl,分别注入液相色谱仪,调节检测灵敏度,使度洛西汀的峰高约为记录仪器量程的20%,分离度试验溶液色谱图中各杂质峰之间分离度应符合要求,系统适用性试验溶液色谱图中度洛西汀杂质F峰与度洛西汀主峰分离应符合要求;精密量取供试品溶液20μl注入液相色谱仪,记录色谱图至度洛西汀主峰保留时间4倍。
但是USP采用的是安捷伦的色谱柱(C8),信息如下:![]()
色谱柱信息:welchrom c18 pn:00310-02041,sn:w132111841 4.6*150mm够清楚了哈?因为没有安捷伦的色谱柱,无图比较,从USP35也可以得到些信息:![]()
![]()
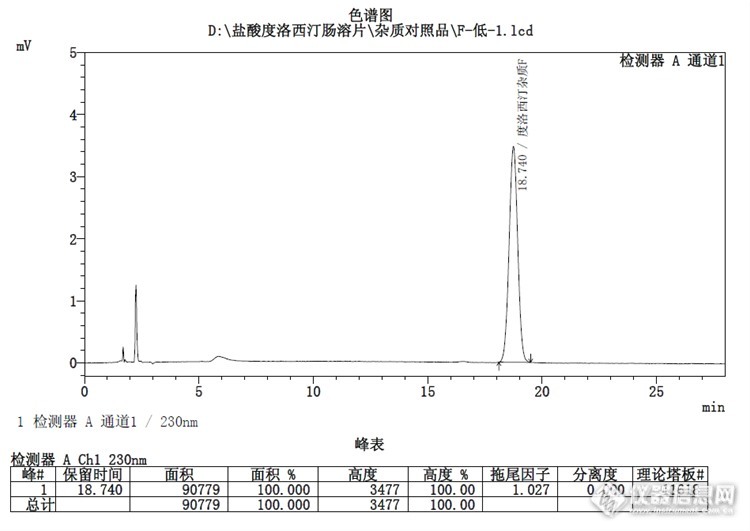
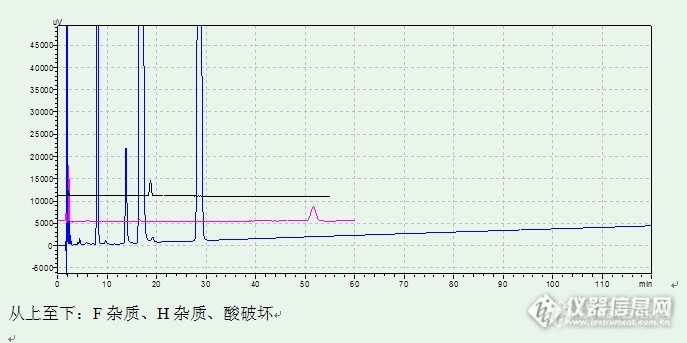
大体情况如上,月旭色谱柱典型色谱图如下:F杂质:![]()
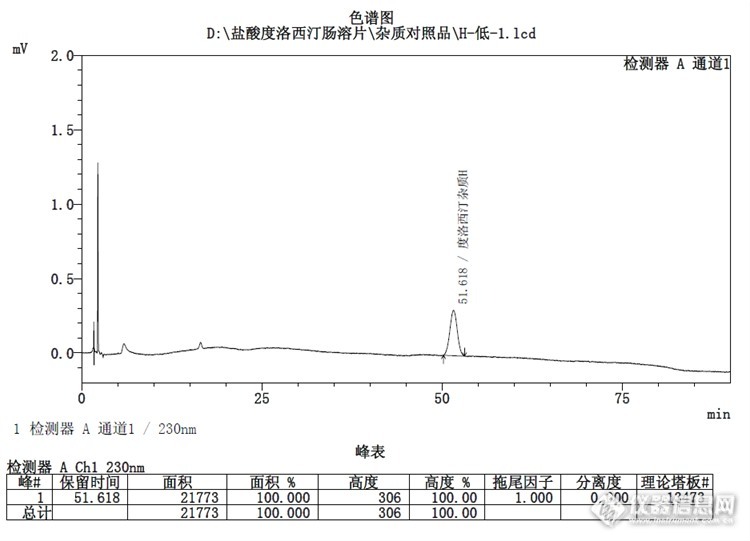
H杂质:![]()
高温破坏杂质:![]()
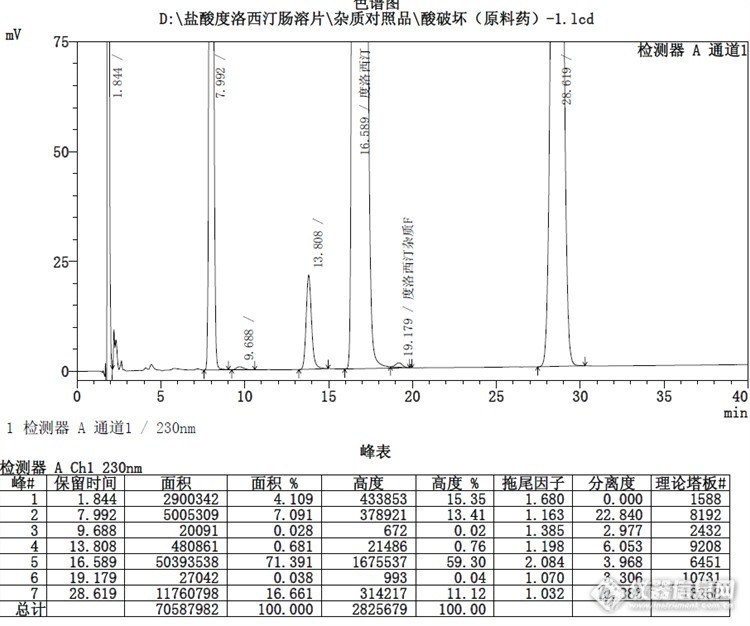
酸破坏:![]()
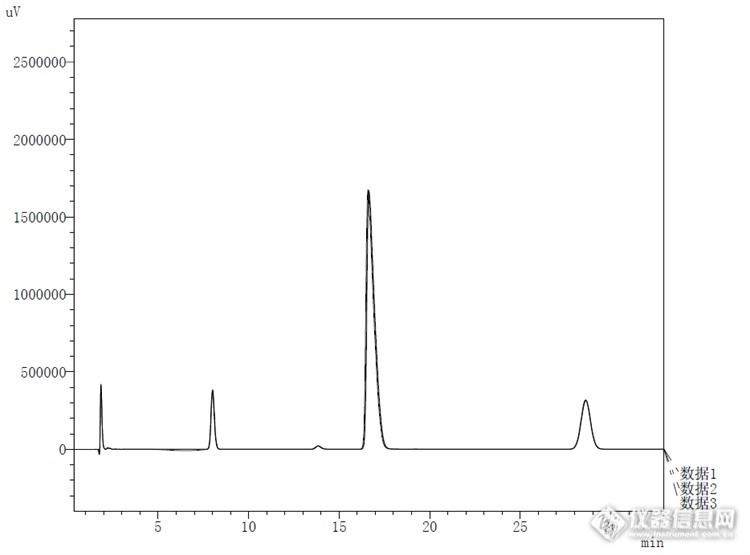
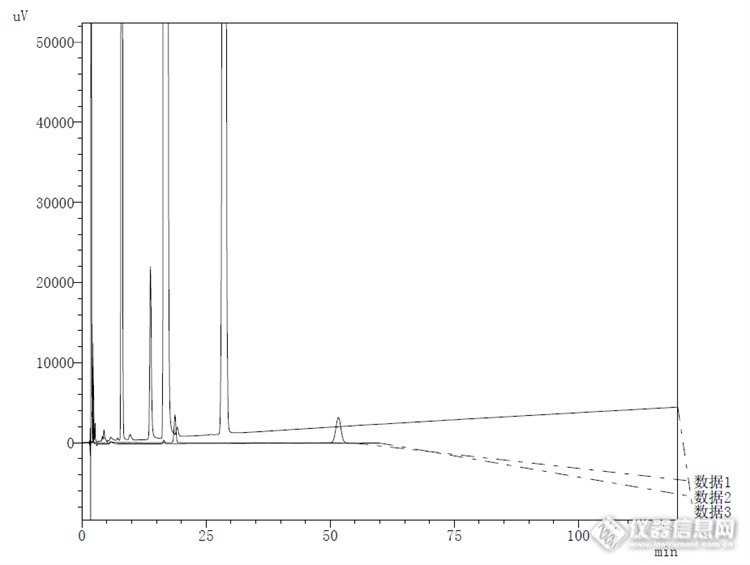
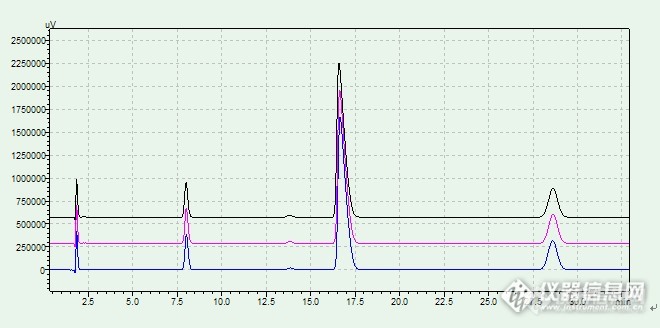
酸破坏对比:连续3针峰重现性较好,说明仪器和色谱柱性能较为稳定。![]()
![]()
清晰对比:![]()
![]()
结论:安捷伦色谱柱暂时还未获得其色谱图,从月旭的C18色谱柱来看完全可以取代。