Establishment of Clean Room Classifications
The design and construction of clean rooms and controlled environments are covered in Federal Standard 209E. This standard of air cleanliness is defined by the absolute concentration of airborne particles. Methods used for the assignment of air classification of controlled environments and for monitoring of airborne particulates are included. This federal document only applies to airborne particulates within a controlled environment and is not intended to characterize the viable or nonviable nature of the particles.
The application of Federal Standard 209E to clean rooms and other controlled environments in the pharmaceutical industry has been used by manufacturers of clean rooms to provide a specification for building, commissioning, and maintaining these facilities. However, data available in the pharmaceutical industry provide no scientific agreement on a relationship between the number of nonviable particulates and the concentration of viable microorganisms.
The criticality of the number of nonviable particulates in the electronic industry makes the application of Federal Standard 209E a necessity, while the pharmaceutical industry has a greater concern for viable particulates (i.e., microorganisms) rather than total particulates as specified in Federal Standard 209E. A definite concern for counts of total particulates in injectable products exists in the pharmaceutical industry (see Particulate Matter in Injections 788 ).
The rationale that the fewer particulates present in a clean room, the less likely it is that airborne microorganisms will be present is accepted and can provide pharmaceutical manufacturers and builders of clean rooms and other controlled environments with engineering standards in establishing a properly functioning facility.
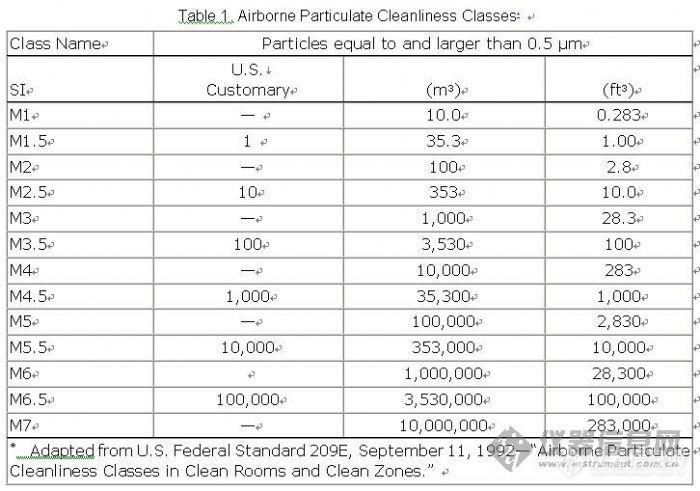
Federal Standard 209E, as applied in the pharmaceutical industry is based on limits of all particles with sizes equal to or larger than 0.5 µm. Table 1 describes Airborne Particulate Cleanliness Classes in Federal Standard 209E as adapted to the pharmaceutical industry. The pharmaceutical industry deals with Class M3.5 and above. Class M1 and M3 relate to the electronic industry and are shown in Table 1 for comparison purposes. It is generally accepted that if fewer particulates are present in an operational clean room or other controlled environment, the microbial count under operational conditions will be less, provided that there are no changes in airflow, temperature, and humidity. Clean rooms are maintained under a state of operational control on the basis of dynamic (operational) data.
![]()