PCR步骤
标准的PCR过程分为三步:
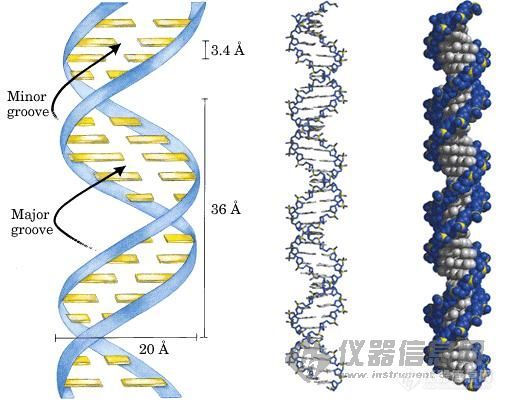
1.DNA变性(90℃-96℃):双链DNA模板在热作用下, 氢键断裂,形成单链DNA
2.退火(25℃-65℃):系统温度降低,引物与DNA模板结合,形成局部双链。
3.延伸(70℃-75℃):在Taq酶(在72℃左右最佳的活性)的作用下,以dNTP为原料,从引物的5′端→3′端延伸,合成与模板互补的DNA链。
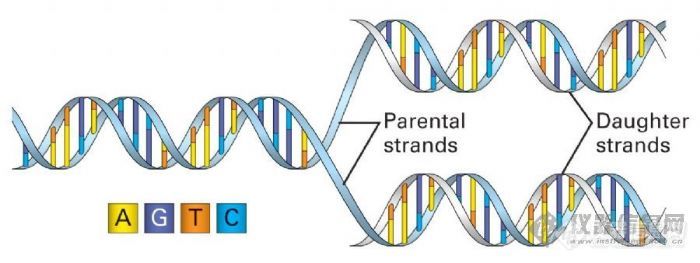
每一循环经过变性、退火和延伸,DNA含量既增加一倍。如图所示:
现在有些PCR因为扩增区很短,即使Taq酶活性不是最佳也能在很短的时间内复制完成,因此可以改为两步法,即退火和延伸同时在60℃-65℃间进行,以减少一次升降温过程,提高了反应速度。
PCR扩增示意图
![]()