有幸参加极限色谱柱的试用活动,分享一下使用结果,希望对大家在色谱柱使用上有点帮助。言归正传。
本次检测目的是监测有机合成反应进程,待测产品本为一个侧链接乌洛托品的化合物(B),原料为含氯代乙酮侧链的物质(A),B在酸性条件下极易水解,在100mmol/L磷酸二氢钠溶液-甲醇(1:1)(用三乙胺调PH=7.0)条件下,一个小时内几乎完全水解成伯胺化合物(C),C由B上的乌洛托品开环所得。本化合物主体为较大的非极性基团,所以色谱柱选择上首选C18柱,分析条件选择有分析结果如下:
仪器:岛津LC-10AT泵、SPD-10A紫外检测器、检测波长:224nm 色谱柱:C18(250*4.6 5um),一根为极限色谱柱,一根为其它品牌色谱柱。
(1)甲醇-水(1:1) 由于易水解,没有调节PH,峰形很差,像一个山包,重复性也差,有时什么峰也不出来,以为是样品分解了,结果临用新配的样品结果也是如此,所以放弃些流动相。把甲醇换成相似洗脱能力的乙腈,结果也是一样。
(2)甲醇-水(1:1)(含2%三乙胺,用磷酸调节PH=7.0) 实验结果与(1)没有明显差别,常常出现的是B与C共存的现象,5分钟内用流动相溶解样品后进样,结果也是一样。
(3)100mmol/L磷酸二氢钠溶液-甲醇(1:1)(用三乙胺调PH=7.0)这个是没有办法的情况下,大大加大了缓冲盐的深度,结果还是存在样品水解的情况。
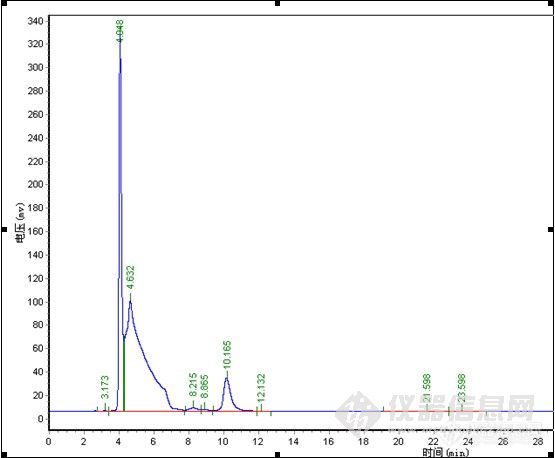
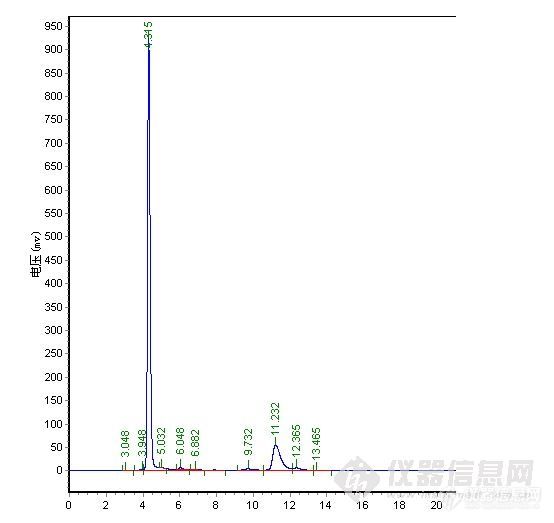
直到现在还没有找到一个重复性好,能很好反应实验进程的流动相,最后经过与合成人员的探讨,得知B完全水解仅会产生C,几乎不存在其它副反应,且高压可能会加速B水解,得知B完全水解仅会产生C,几乎不存在其它副反应,最后决定直接使B转化为C后再检测也不失为一个好办法,C性质稳定,容易检测。以下是在流动相(3)下得出的谱图。
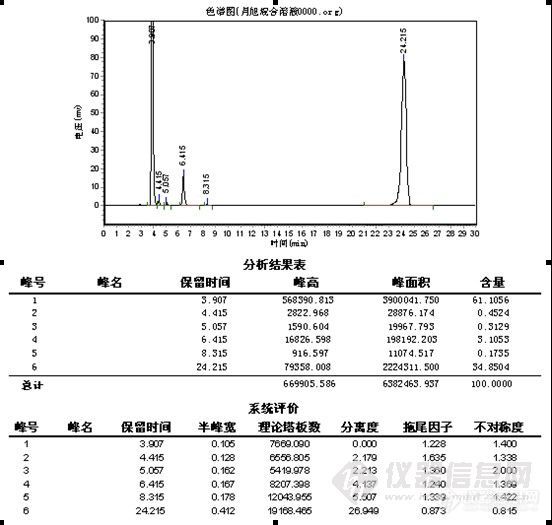
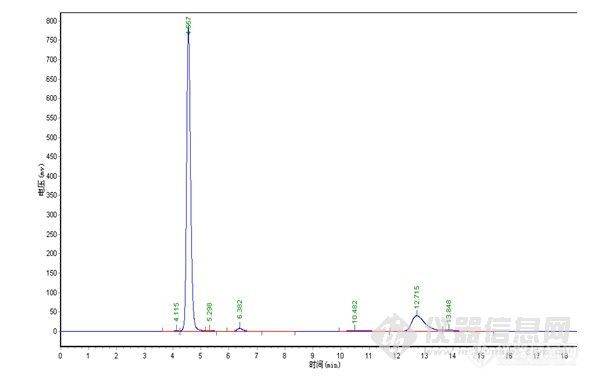
极限色谱柱:
![]()
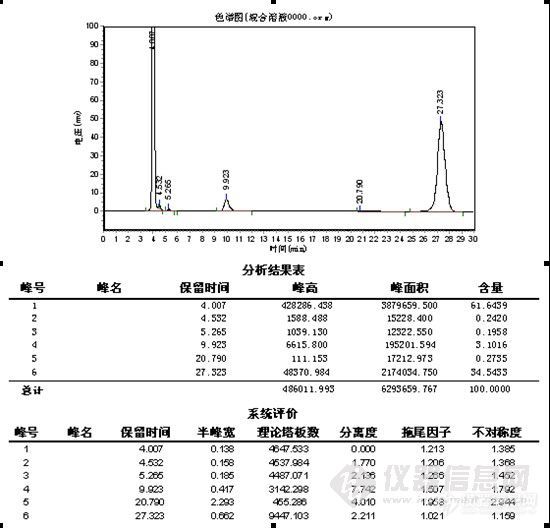
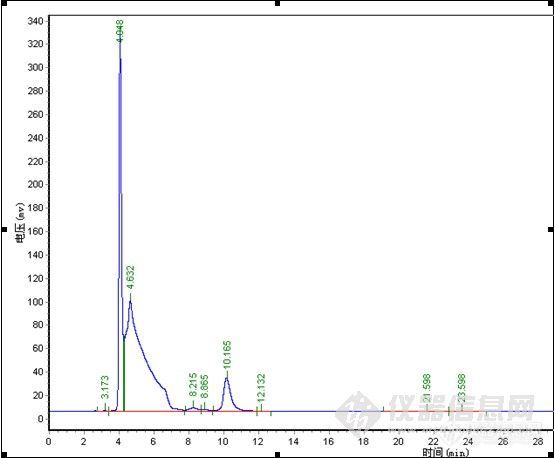
其它品牌:
![]()
总结:对比中,在纯甲醇 1.0ml/min下,极限色谱柱压为5.8MPa,其它品牌色谱柱压为6.7MPa,加有保护柱,可能会略高于其它用户。4.0min为物质C,要完全水解下,未见B色谱峰,24.2min\27.3min为A,理论塔板数、分离度明显极限色谱柱较高,分析时间较短,但在峰形上,特别是A物质来看,极限色谱柱没有体现出优势,不过从A物质的峰形没有托尾上看,极限色谱柱在封端技术上有其独到之处。
本实验试用的三种流动相非是最佳方案,后述会继续了解极限色谱柱在酸性条件下的表达,其使用寿命也是一个重要的考察项目。前几年也恰好参与过极限色谱柱的试用活动,检测的物质为18种氨基酸注射液,方法为柱前衍生,使用效果还不错,有做这一方面的版油可以一试。