获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 amugong(amugong) 发表:谢谢。不过我试验过了我这个物质不溶于水。我试验了下溶的最好是的丙酮和DMSO,其他溶剂例如甲醇,乙腈均可以溶解一部分,但是溶解度不高。我这个化合物据报道应该亲脂性较强,有四个酚羟基和两个外侧苯环。内部是个疏水性结构。现在通过加大有机相比例和流速基本改善了峰型,目前又遇到的情况是几十针进样后发现有残留。进样前的空白样品是没有的,进样以后,再进空白开始出现较高的残留。基本排除进样针污染,是色谱柱残留。您对于这种情况有什么改善的经验吗?
羟基多,极性大,C18柱一般要加酸会使峰形和保留都有改善,但[url=https://insevent.instrument.com.cn/t/Yp]液质的负源加酸响应会变低,加乙酸铵也会使峰形变好,hilic出峰如果慢,水相比例加大即可,浓度到10-20mm试一下。
原文由 syzhudi(v2952102) 发表:你能查到这个物质相关的物化性质么?熔沸点啥的,不行的话用气质试试。当然还有一种方法,把管路全部换成金属,然后用正相色谱走。。。
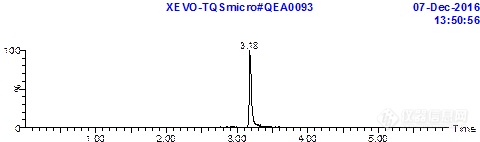
增加有机相比例是有效果的。我把95%的高有机相改到100%,流速加大。得到了下面的图,明显改善了拖尾。但是又有了新问题,这个梯度洗不干净柱子上的目标化合物,影响定量。我这个化合物是个柱前衍生得到的产物。没有标准品,文献里也没有具体说明,实验结果应该显示极性比较小的,我试了下很多有机试剂都能溶蒸干的固体,但是溶解性最好的是DMSO和丙酮,异丙醇或者极性再小点的乙醚,己烷都溶不好,甲醇比乙腈溶解性稍微好一点。我现在用的是ACN的流动相,换成甲醇响应会高5倍左右,但是峰型很差,同样洗不干净。。进样针已经设置成进样前后各用纯甲醇洗10s,空白样品仍然有较高的残留。我判断应该是色谱柱残留。我想问下对于这种亲脂性,弱酸性的物质(带两个苯环),有更合适的色谱柱来分离吗?
原文由 东海710(v2945599) 发表:查了好久,很难查到有用的信息。我这个物质是个柱前衍生物,相关资料极少。有的也只是一笔带过,说是性质非常稳定,难溶于大部分有机溶剂,不溶于水。工业应用也只是作为一个中间产物来制备其他化合物,用的时候只是用一些有机溶剂重悬后,再用其他试剂进行处理,再反应。我试验了下应该微溶于很多有机溶剂,有上核磁打结构的,用的是氘代氯仿,我估计氯仿应该是可以溶解的。因为有颜色,微量的蒸干物我用甲醇超声以后还是可以溶的比较干净的。正相不太现实,对泵也不好。算了,还是正常条件下再改进吧。本来想放弃了,但是查了很多资料原来还没有人用[url=https://insevent.instrument.com.cn/t/Yp]液质做过这个东西,觉得还是好好摸索条件把这个事情搞定吧。液相一般都是走60min的梯度,样品多的话太折磨人了。哎,再试试再试试
你能查到这个物质相关的物化性质么?熔沸点啥的,不行的话用气质试试。当然还有一种方法,把管路全部换成金属,然后用正相色谱走。。。