维权声明:本文为xx_dxd_xx原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
1 引言 银杏叶提取物是目前临床广泛应用的天然产物药。西方学术界对该药物的成分和疗效也广泛接受。目前银杏叶提取物执行的标准基本上都源自德国EGB-761,其中除规定有效成分含量外,还对银杏酸含量进行了限定。按欧盟药典【European Pharmacopoeia 8.0.Monograph: Ginkgo dry extract. Refined and quantified.
2008】、美国药典【United States Pharmacopeia.Thirty-seventh Revision: Powdered Ginkgo Extract.
2015.】要求,总银杏酸含量不得超过5 mg/Kg。我国执行的标准略为宽松,2015版药典【中华人民共和国药典
2015, 一部:416-417页】规定总银杏酸含量不得超过10 mg/Kg。
.
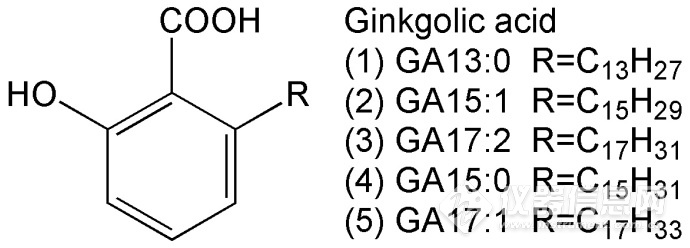
![]() 图1银杏酸的结构式
图1银杏酸的结构式
. 银杏酸的分子结构如图1其中GA13:0、GA15:1、GA17:2是药典规定需要检测的项目。银杏酸的色谱分离并无难点,但是要测定银杏叶提取物中的银杏酸却存在两方面的问题:一是银杏酸限量很低,几乎接近仪器检出限;二是体系中共存的黄酮、内酯、糖类、蛋白质等成分复杂,基体干扰严重。目前通行的检测方法有欧盟药典法、中国药典旧版(2010版)方法、中国药典2015版方法,对比见表1。
![]()
色谱分离方面,各种方法均采用反相色谱,条件略有差异,但无本质不同。提取方面,欧盟药典采用甲醇溶剂,提取较为完全,但共存的大量极性组分也一起提取进入溶液中,形成严重的基体干扰。而我国2010版药典方法采用石油醚提取,极性组分溶解少、干扰有所降低,但石油醚对极性试样的浸润性和渗透性不好,提取不完全,因此出现结果偏低的现象。相关研究人员【姚建标, 等.药物分析杂志,
2015, 35 (11): 2041-2044.】借鉴了欧盟药典的提取方法提出了2015版药典方法,但是将试样量从0.5g增加到2g。增加样品量是为了弥补检测灵敏度的不足,因为2015版药典方法采用的紫外检测波长为310nm,这一波长下银杏酸的吸收强约为210nm下的五分之一。当然,在310nm下共存杂质的吸收更小、色谱基线漂移的问题也有所缓解,但是取样量增大使得更多的基体杂质进入色谱柱,基体干扰问题有增无减。
.
众所周知,通过液液萃取、固相萃取等前处理手段可以起到减少基体干扰、富集目标物的作用,相关研究也有一定报道【Stefan U, Iuliana D S, Victor D,Andrei M.
J. Liq. Chromatogr. R. T.,
2010, 33 (1): 133-149. Ji W, Ma X, XieH, Chen L, Wang X, Zhao H, Huang L.
J.Chromatogr. A,
2014, 1368 (1):44-51.】。但是这些前处理过程较为繁琐,不利于推广。也有通过超高效
液相色谱-串联质谱方法进行银杏叶提取物中银杏酸的检测【孙健, 等. 色谱,
2016, 34 (2): 184-188.】,灵敏度和基体干扰问题都能较好的解决,但仪器昂贵,也难以推广。
.
本课题组在进行银杏活性成分提取研究的过程中也遇到了上述测试问题。为了改进现有方法,我们将简便有效的“分散液液微萃取”方法应用到样品的前处理过程中,有效解决了基体干扰问题,灵敏度也显著提高,同时该前处理方法十分简便,样品和试剂的用量也很少。相关研究前沿和创新内容已经撰文投稿,现将较为基础的实验方法介绍如下。.
2 实验方法
2.1 仪器与试剂 大连依利特230型高效
液相色谱仪,安捷伦HC-C18(4.6*150 mm,5 μm)色谱柱。
色谱纯甲醇、乙腈为上海国药产,纯水为石英亚沸蒸馏。萃取剂为ACS级三氯乙烯,其余试剂为分析纯。
银杏叶提取物试样购于湖北某公司,相关制剂为市售药品(天保宁、金纳多)。
混合银杏酸标准品购于武汉某公司,总含量不低于99%,其中GA13:1占10.1%,GA15:1占50.5%,GA17:1占34.7%,GA17:2占1.7%,GA15:0占3.0%。用甲醇溶解配置成总银杏酸浓度为1.000 g/L的标准储备液,4℃保存。使用时用甲醇稀释成所需浓度的工作标液。
.
2.2 实验方法
a)提取
准确称取0.5 g试样,加10 mL甲醇,常温超声提取10 min,冷却后定容、摇匀。
上述溶液用微孔滤膜过滤,收集滤液。
b)萃取
取上述滤液3.00 mL移入尖底离心管中,加入萃取剂三氯乙烯0.150 mL混匀,此时不分层。
加入水溶液(含10%无水硫酸钠 + 0.2%三氟乙酸)7.00 mL,加塞、摇匀,此时应得到浑浊液。
4000转离心3min,三氯乙烯萃取剂沉到底层。
c)取样
用注射器吸取三氯乙烯层0.100 mL移入样品瓶,氮气吹干,加0.100mL甲醇溶解。
d)色谱测定
条件见2.3。
.
2.3 色谱条件 采用Agilent HC-C18(4.6*150mm,5μm)色谱柱,甲醇-水-三氟乙酸体系(体积比92:8:0.1)为流动相,流速1.00mL/min,检测波长242nm,进样量50.0μL。
.
2.4 定量分析 银杏酸工作标液直接进样测定,建立工作曲线,外标法定量。计算待测样时需乘以萃取的富集因子。富集因子为20。
.
3 实验结果
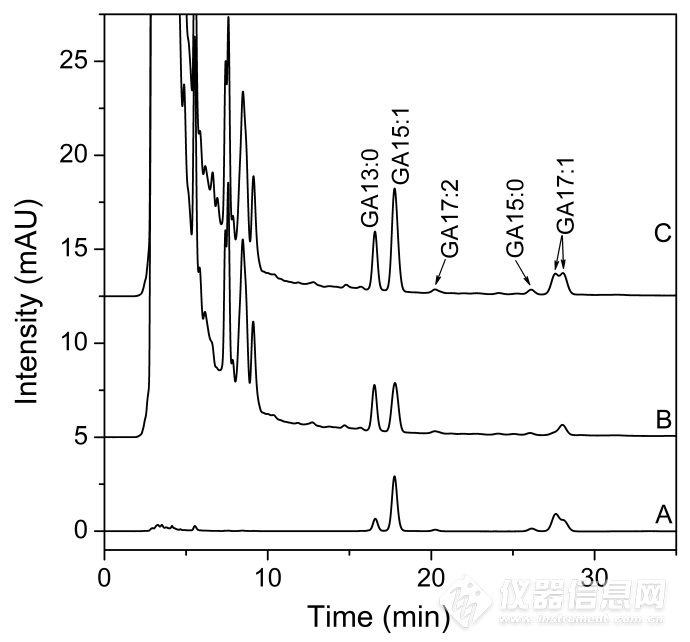
3.1 色谱图 银杏酸标样、银杏叶提取物试样、试样加标的色谱图见图2,分离效果较好,基线平直、基体干扰小。
![]()
.
3.2 方法验证 将银杏酸的甲醇标液按试样同样方法萃取和检测,考察了方法的重现性、线性范围、定量限(按S/N=10计算),测定了萃取过程的萃取率和富集因子,结果见表2。方法重现性2.9% ~4.8%。方法定量限0.007 ~ 0.020 mg/L,测定上限为5.00 mg/L。按取样0.5g定容至10mL计算,对试样的定量限为0.14 ~0.40 mg/Kg,对试样的测定上限为100 mg/Kg。
萃取率达到95.4% ~98.3%,富集因子达到19.1 ~19.7。
![]() 3.3 样品测定
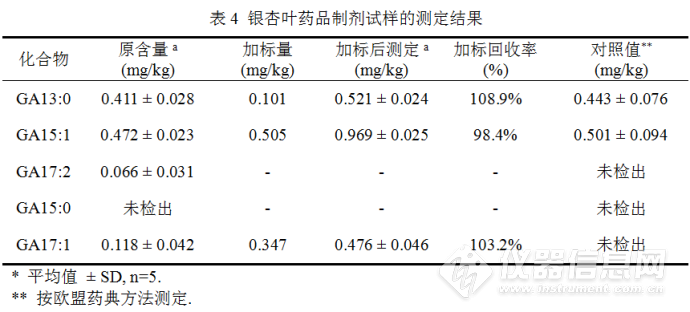
3.3 样品测定 银杏叶提取物试样的测定结果见表3,进行了加标回收实验(总银杏酸加标量为5.00mg/Kg),加标回收率94.0% ~ 100.8%。以欧盟药典方法进行对照试验,结果与本方法一致。药品制剂试样的测定结果见表4,同样进行了加标回收实验和对照实验,结果也较好。
![]()
![]()
.
4 相关讨论 (1)试样体积与萃取剂体积之比影响萃取率和富集因子。此比值大时富集因子高但萃取率明显低于100%。反之则导致萃取率接近100%但富集因子很低。本方法选择3.00 mL试样加0.150 mL萃取剂,是兼顾两个方面考虑的结果。此时萃取率达到95.4% ~98.3%,接近100%。富集因子达到19.1 ~19.7,接近体积比计算结果,因此在定量计算时近似认为富集因子为20。
(2)萃取剂要选择低沸点、高密度、弱极性、难溶于水的有机溶剂,符合条件的主要是卤代烃。对比表明三氯乙烯效果最好。
(3)试样溶剂为甲醇,与萃取剂能够互溶,不能分成两相。但加入适量水之后可以实现分层。极性强的基质进入水相,从而使干扰减小。极性弱的银杏酸进入三氯乙烯层实现富集。水中加入10%硫酸钠起盐析作用,加0.2%三氟乙酸可以抑制银杏酸电离,都有利于提高萃取率。不加时萃取率显著降低。
(4)萃取过程形成浑浊的乳状液,两相接触面积大,因此很快达到平衡。加水溶液后震荡30秒到1min即可。萃取时间对结果几乎不影响。
(5)萃取在室温下进行,温度变化对结果影响不大。
(6)银杏酸的紫外吸收有3个特征峰,分别为210 nm、242 nm、310 nm。使用210 nm虽然灵敏度高,但干扰大。本方法使用242 nm检测,实际信噪比高于210 nm。使用310 nm检测的信噪比略有降低。
(7)由于萃取减少了基体干扰、实现了富集,本方法使用等度洗脱进行色谱分离也能获得很好的效果,并且没有基线漂移。本方法灵敏度也显著由于药典方法,定量限达到0.14 ~0.40 mg/Kg,而药典方法的检出限为0.5 mg/Kg。
.
5 结论 采用“分散液液微萃取”方法对银杏叶提取物样品进行前处理,具有简单快捷的优点,同时减少基体干扰、实现富集的效果较好,能有效提高信噪比,降低检出限和定量限。方法准确可靠,易于推广。