维权声明:本文为byron1111原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
SH 0806-2008 中间馏分芳烃含量的测定 分析方法的注意事项----流动相
概述:SH0806-2008标准使用高效液相色谱方法测定柴油和馏程范围为150-400℃的石油馏分中单环芳烃、双环芳烃、三环以上芳烃和多环芳烃含量。该分析方法虽然比较简单,但是分析条件的控制比较重要。在实验操作中,尤其需要注意流动相的问题,流动相不良会造成保留时间漂移从而使得色谱柱切换时间选择发生困难。一 原理介绍
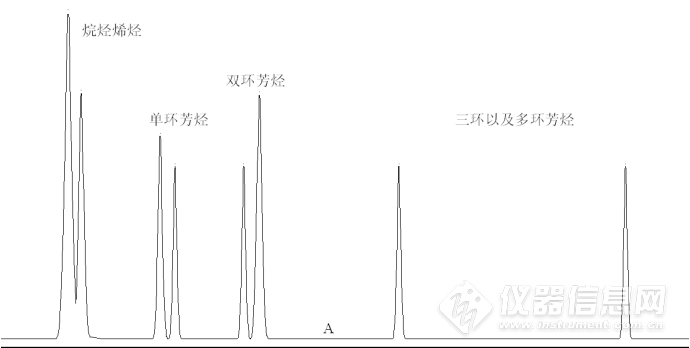
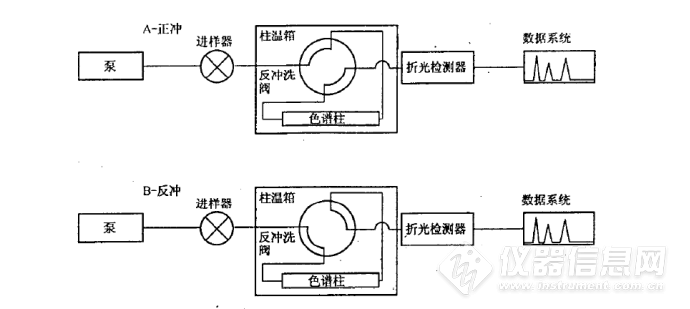
系统结构原理如图1所示,系统使用正庚烷做流动相,单输送泵、极性色谱柱(氨基或者氰基柱)、四通阀和示差检测器实现分离。待机状态和进样状态下(即状态A),流动相自右向左流过极性色谱柱,柴油中的烃类和芳烃类组分实现分离。理想情况下,单环芳烃、双环芳烃、多环芳烃依次在色谱柱出口流出,如图2所示。![]()
图1 SH 0806 方法的硬件原理图
![]()
图2 色谱柱内样品理想分离示意图
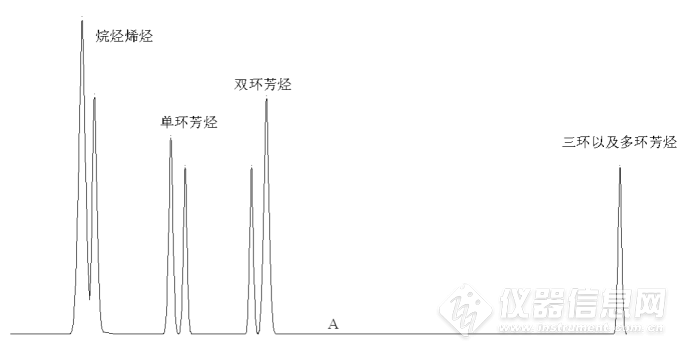
当双环芳烃流出色谱柱后(色谱图中A点位置,或称为切换点),四通阀旋转,系统状态变为反吹。色谱柱内流动相的方向变成自左至右,将三环以及三环以上的芳烃类物质反吹出色谱柱,在示差检测器上表现为单峰,如图3所示。![]()
图3 最终谱图
二 分析注意要点
首先要注意该方法的原理是比较理想化的,干扰因素也比较多。柴油中的单环芳烃、双环芳烃、多环芳烃是否可以清晰彻底的分离开,是难以保证的。况且柴油中的二烯烃、杂环类、酯类化合物等都会对分析结果带来影响。其次,色谱柱的选择十分重要,具体的选型需要咨询色谱柱厂家。再次,切换点的选择非常重要。样品组成可能比较复杂,A点可以选择的时间窗口就会比较窄,需要多次重复实验寻找合适的切换点。分析条件需要非常稳定,需要较为严格的控制流动相组成、泵输送流速以及色谱柱温度,以免影响保留的重复性。三 常见问题——流动相
保留时间的漂移是最为常见的问题。在进样系统性能测试标准样品时,芳烃的保留时间长时间的漂移,致使难以确定切换点。往往会耗费较多时间来平衡系统,等待保留时间稳定,从而降低分析效率。其本质的原因在于流动相的不稳定。笔者曾经在使用该系统时,长时间重新系统后,连续进样10余次系统测试标样,发现芳烃的保留时间不断发生缩短,认为色谱柱未彻底平衡。第二天更换了流动相(新换的流动相没有彻底封口放置在实验室10小时),进样3次,芳烃的保留时间较为稳定。后来考虑了一下,原因应当为新换的正庚烷中水含量已经与空气中的水含量交换平衡,或者说水含量已经比较稳定,进而使得芳烃保留稳定。四 小结
SH 0806-2008系统分离原理属于正相液相色谱,我们知道正相HPLC一般不太容易得到良好的保留时间重复性。原因是在正相HPLC分析中,流动相中的微量水会显著的改变其极性。假设流动相原先的极性为0.01,吸收微量水之后极性变为0.02,看上去似乎变化不大,但其实极性增大了一倍。尤其是使用硅胶色谱柱的场合,流动相与环境空气中的水蒸气发生交换,改变了极性,从而影响保留时间。在使用硅胶柱分析时,一般要避免使用彻底干燥的正己烷流动相,避免吸水造成保留不稳定,甚至需要特意在流动相中加入微量的水。