维权声明:本文为v3255306原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
沉淀蛋白法结合HPLC-MS/MS测定右佐匹克隆在人体血浆中的含量及药物动力学应用
摘要
失眠症是一种持续性的身体疾病,睡眠质量或数量都不令人满意。右佐匹克隆(E-ZOP)是一种人工合成的治疗失眠的药物,能有效地维持睡眠质量。本研究建立了一种高效、快速的高效
液相色谱-串联质谱(HP
LC-MS/MS)测定人血浆中E-ZOP的方法,并应用于药代动力学研究。该方法避开了常用的液-液萃取法或固相萃取法,而是用甲醇和二甲基亚砜一步沉淀法进行样品制备。高效
液相色谱-质谱/质谱联用技术已完全用于人血浆中E-ZOP的测定,并成功应用于人体药代动力学研究。口服E-ZOP后,峰值时间(Tmax)为0.924 h,半衰期(T1/2)为4.902 h,在人体内表现为快速吸收、缓慢消除,动力学呈线性变化。本研究首次提供了一种高效的E-ZOP蛋白沉淀提取方法,在人体药代动力学研究中显示出明显的优越性。
关键词:高效
液相色谱-质谱联用;艾司唑匹克隆;蛋白沉淀;药代动力学;
1.简介
失眠已被强调为主要的公共卫生问题[1,2]。睡眠问题的高发生率和广泛的对精神和身体健康的影响[3]和身体健康[4],以及功能受损[5],使得人们越来越意识到获得足够高质量睡眠的重要性[6]。E-ZOP用于治疗不同类型的睡眠障碍[7,8],研究表明,它是由其GABA受体复合物与苯二氮卓受体[9]耦合而成,通过激活GABA受体增加神经抑制,具有明显的镇静作用[10]。E-ZOP(化学名为(S)-(+)-6-(5-氯-2-吡啶基)-7-氧代-6,7-二氢-5H-吡咯[3,4-b]吡嗪-5-基-4-甲基-1-哌嗪羧酸盐,是一种非苯二氮卓类催眠剂,属于环吡咯啉类[11],微溶于水/乙醇,溶于磷酸盐缓冲液(pH 3.2)[12,13]。Gary K Zammit[14]评估了E-ZOP的药代动力学,结果表明,R,S-佐匹克隆的清除率是立体选择性的,与活性较低的R-对映体相比,E-ZOP的清除率更低,T1/2更长。在这几十年中,研究发现,受试者在夜间服用3 mg的E-ZOP,第二天早上他们的精神运动功能不同程度地降低,这可能导致诸如驾驶、记忆和协调等不可察觉的活动的减少[15]。为了更好地了解E-ZOP的药代动力学,需要一种灵敏、特异、准确的测定人血浆中E-ZOP的方法,以观察E-ZOP的量效关系。
关于使用高效
液相色谱-紫外色谱法(HPLC-UV)、
气相色谱法(
gc)、
LC-MS/MS和高效薄层色谱法(HPTLC)对生物液中E-ZOP进行定量的论文已经发表[12,16]。N Sharma等人[17]报道2013年采用LC-UV方法在13分钟内定量E-ZOP,曲线范围为0.02-7.2 μg/mL,但分析时间太长,无法实现高通量[18,17,19-22]。Van Bocklaer等人[23]报道了首次采用
气相色谱-串联质谱(
gc-MS)测定血液和胃内容物中E-ZOP的方法,方法检测E-ZOP的最低定量限(LLOQ)为2 ng/mL[24],与
LC-MS/MS相比,该方法的灵敏度较低。Gebauer MG等人[22]报道开发了液-液萃取(LLE)相结合的液-液萃取(LLE)
LC-MS/MS方法[22,25,26-29],该方法仅使用50 μL样品体积,E-ZOP的LLOQ为0.1 ng/mL。Meng等人报道了[15]
LC-MS/MS法与固相萃取法(SPE)联用测定人血浆中E-ZOP的方法,曲线范围为1.00–100 ng/mL,使用50 μL样品体积是该方法的亮点。在这些技术中,
LC-MS/MS的灵敏度最高,这是由于其具有较高的选择性。然而,包括LLE和SPE在内的样品预处理过程相对复杂,例如,用温和的氮气流分离离心后的有机层并在40 ℃下蒸发干燥[30-36]。这种预处理不仅增加了操作的复杂性,降低了处理量,而且还导致了不良的有机污染,这是临床实验室难以接受的。因此,在药代动力学研究中,E-ZOP含量测定中存在的样品制备问题亟待解决,本文提出了一种新的蛋白质沉淀法-高效
液相色谱-质谱/质谱联用技术,从特异性、线性、灵敏度、准确度、精密度、基质效应等方面对E-ZOP在人血浆中的测定进行了充分验证,回收率和稳定性,最终应用于口服给药的人体药动学研究,并用DAS 3.0软件通过无室模型计算药动学参数。结果表明,E-ZOP在人体内吸收快,消除慢,动力学呈线性。定量下限(LLOQ)为0.1 ng/mL,R2>0.99为线性曲线。在0.1~30 ng/mL范围内测定,日内和日间精密度分别小于9.99和8.71 %,平均准确度为95.53 %~109.18 %。该方法回收率高,基质效应可忽略。E-ZOP的药代动力学参数包括Cmax =13.273±2.717 ng/mL,Tmax =0.924±0。446h,AUC0−t =69.308±14.895 ng/mL•h,T1/2 =4.902±0.819 h。本研究用氘化E-ZOP代替卡马西平作为稳定同位素标记内标(SIL-IS)来定量E-ZOP。本文介绍了一种优化的高效
液相色谱-质谱/质谱法测定人血浆中E-ZOP的方法,并成功应用于E-ZOP片的临床药代动力学试验。
2. 实验
2. 1 材料与试剂
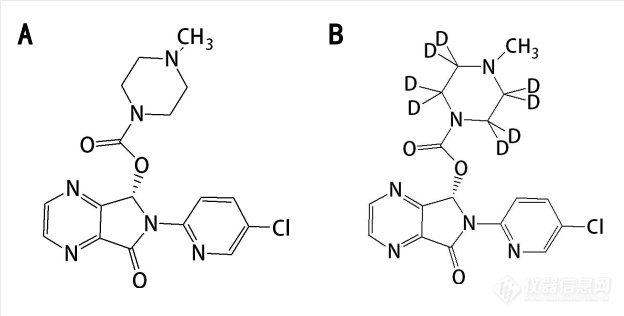
E-ZOP购自中国食品药品检定研究院(纯度>99.8 %)(图1),批号:19120474。E-ZOP-d8(纯度:98.7 %)从多伦多研究化学公司采购,批号:11-PTR-48-1(图1)。甲醇(
LC-MS级)从Fisher Chemical购买,流动相的所有水溶液均采用D11951超纯水机(德国赛默飞世尔)制备。甲酸(HPLC级)从MREDA采购,批号095224。醋酸铵(HPLC级)从MREDA采购,批号015966。二甲基亚砜(分析级)购自国药集团化学试剂有限公司,批号20180622。所有其他化学品和试剂均为分析级。
2. 2 设备和操作条件
2. 2. 1
液相条件
分析采用HPLC/MS/MS系统进行,该系统由资生堂HPLC系统(日本)和AB Sciex API 5500串联质谱仪(美国应用生物系统公司)组成。在C18柱(资生堂CAPCELL PAC MGⅢ,2.0×150 mm,5μm)上用甲醇(10 mM醋酸铵,0.1%醋酸):水(10 mM醋酸铵,0.1%醋酸)=55:45的流动相,流速为0.35 mL/min,实现了E-ZOP和E-ZOP-d8的分离。将样品注入5 μL等分试样中。
2. 2. 2 质谱条件
采用配有电喷雾电离(ESI)源的API 5500串联质谱仪(Applied Biosystems SCIEX,America)对HP
LC-MS/MS分析进行多反应监测(MRM)检测。每一个前体离子经过碰撞诱导的离解,以确定产生的产物离子。以高效
液相色谱(HPLC)为流动相,通过注射泵,优化了E-ZOP和E-ZOP-d8溶液的界面无关仪器参数。2. 3 储备溶液和工作溶液的制备
储备液用甲醇溶解精确称量的标准对照品,E-ZOP的最终浓度为1000 μg/mL。将精确体积的0.10 mL E-ZOP标准溶液移入10 mL容量瓶中,并用甲醇稀释至一定体积,以进一步稀释E-ZOP储备液取10 μg/mL的工作液,用甲醇稀释,得到2、4、10、20、100、200、400和600 ng/mL的工作液。同时,将精确重量的10.00 mg E-ZOP-d8原料药转移到10 mL容量瓶中,然后溶解并用纯品稀释至一定体积甲醇制备1000μg/mL的储备液。E-ZOP-d8的工作液浓度为50 ng/mL。所有药物的储备液在4 ℃的避光容器中储存至少60天,无变化。
2. 4 校准标准和质量控制(QC)样品的制备
在0.1、0.2、0.25、0.5、1.0、2.5、5.0、10、20、25、30 ng/mL血浆中制备E-ZOP校准标准。根据FDA关于选择QCs的指南[36],为了进行准确度和精密度研究,QCs在4个浓度水平下制备为6个重复品,包括定量下限(LLOQ)、低(L:定义为LLOQ的三倍)、中(M:定义为中范围)和高(H:定义为高范围)。而对于其他试验(在志愿者样品分析期间),只使用了3种浓度水平(LQC、MQC和HQC)的复制品。对于E-ZOP,分别在0.1、0.25、2.5和25 ng/mL下制备LLOQ、LQC、MQC和HQC。
2. 5 样品准备
将190 μL对照人血浆置于2.0 mL离心管中,加入精确体积的10 μL 2-600 ng/mL工作溶液,以获得E-ZOP的0.1-30 ng/mL血浆浓度。添加以下25 μL的IS(50 ng/mL)。然后加入300 μL沉淀蛋白溶剂(MeOH:DMSO=7:3,0.1%甲酸)振荡10 min提取E-ZOP和E-ZOP-d8,在4 ℃下以14000 rpm离心15 min分离上部有机相和下部水相。将100 μL上清液溶解于200 μL水相中,并涡旋混合2分钟。将5 μL等分溶液注入
LC-MS/MS系统进行分析。
2. 6. 方法验证
2. 6. 1 专属性
通过比较六个人的空白血浆样品、在最后给药后0.5小时从其中一名受试者获得的临床血浆样品、以30 ng/mL的剂量加入E-ZOP的血浆样品和以0.1 ng/mL的剂量加入E-ZOP的血浆样品的色谱图来评估特异性和内源性干扰。通过比较50 %甲醇和添加E-ZOP的三蒸馏水(0.1 ng/mL)的LLOQ和Esopiclone-d8(50 ng/mL)的色谱图来评估特异性和外源性干扰。制备并分析所有空白血浆样品,以确保没有干扰峰。
2. 6. 2 最低定量限(LLOQ)
采用1/X2加权线性最小二乘回归模型,以E-ZOP/E-ZOP-d8与血浆浓度的峰面积比构建校准曲线。LLOQ表示线性范围内分析物的最低浓度,可以用可接受的精度和准确度进行测定。
2. 6. 3 精密度和准确度
在同一天对四种浓度(0.1、0.25、2.5和25 ng/mL)的六个重复的LLOQ和QC样品进行分析,以评估日内精密度和准确度。通过连续三天分析LLOQ和QC样本来评估日间精密度和准确度。方法的精密度和准确度分别用相对标准偏差(RSD)和相对误差(RE)表示。RSD和RE均不得超过15 %。然而,在LLOQ,RE和RSD<±20 %是可以接受的。
2. 6. 4 基质效应与提取回收率
提取回收率是通过比较在6个不同批次血浆中制备的三个水平的QC样品中提取的分析物与IS比的绝对峰面积与空白血浆、高溶血性血浆和高脂肪血浆提取后用相同浓度的分析物纯溶液进行强化LQC、MQC、HQC。通过比较六批样品在LQC、MQC、HQC水平下强化的空白血浆提取物与相同浓度水平分析物强化的空白水提取物中分析物绝对峰面积与IS比值的比较,来评估基质效应。
2. 6. 5 稳定性试验
对三种不同浓度的QC样品在不同条件下的稳定性进行了分析:(1)连续三次冻融循环(从-20 ℃到23 ℃);(2)室温(23℃)制备前3h;(3) 冰箱温度(4 ℃)制备后20小时,室温(23 ℃)制备后6小时;(4)自动进样器10 ℃制备后24小时;(5)冰箱温度(−20 ℃)制备前3、8、31天。通过比较储存的QC样品和新制备的样品的平均浓度来评估溶液的稳定性。样品被认为是稳定的,与标称浓度的偏差在±15.0 %以内。所有稳定性试验样品在6个重复中进行分析,并根据新制备的样品确定偏差。
2. 7 药代动力学研究的应用
对12名健康男性受试者进行了药代动力学研究。伦理委员会批准了方案,志愿者得到了知情的书面同意。给药前禁食12小时,给药后禁食3小时。口服给药后,在给药前和0时从颈静脉抽取2 mL的血样。后分别于0.083,0.167,0.333,0.5,0.75,1,1.25,1.75,2,2.5、3、4、5、6、8、12和24小时取血。实验期间,可以自由饮水。随后在14000×g转速下离心10分钟制备血浆,并在-80 ℃下立即冷冻。
2. 8. 数据分析
药代动力学分析采用DAS2非房室模型软件程序(中国数学药理学专业委员会,中国上海)计算AUC、Cmax、Tmax、T1/2和CLz/F。数据以平均值±SD表示。
3. 结果与讨论
3. 1. 方法优化
3. 1. 1. 色谱条件优化
为了获得满意的色谱行为和最大限度地提高E-ZOP和IS的电离响应,我们在流动相体系上进行了几次尝试。由于E-ZOP具有酸性,故采用醋酸溶液作为流动相,以提高响应速度。考虑到流动相pH范围的稳定性和消除色谱峰的分裂,在流动相中加入了10mM乙酸铵。以不同比例的甲醇-水和乙腈-水对E-ZOP进行洗脱实验,发现甲醇比乙腈具有更低的背景噪声和更好的分辨率。根据峰形、保留时间、稳定性和灵敏度,甲醇(10 mM醋酸铵,0.1%乙酸)-水(10 mM醋酸铵,以0.1 %乙酸(55:45,v/v)为流动相。资生堂CAPCELL PAC-MGⅢC18柱对E-ZOP有较好的保留作用,10 mM乙酸铵对E-ZOP的反应显著增强。在优化的高效
液相色谱条件下,检测到E-ZOP和E-ZOP-d8(IS)在保留时间为1.97和1.90分钟。总运行时间是4.5分钟。
结合E-ZOP在水和乙醇中微溶、在磷酸盐缓冲液(ph 3.2)中微溶的特点,Shaikh K[16]利用0.05 M一元磷酸钠缓冲液组成的流动相,建立了一种灵敏的E-ZOP定量方法。N Sharma[15]提出了一种使用0.01 M磷酸盐缓冲液和正磷酸的UPLC方法。在PKa=6.89的条件下,我们设计了一种pH=4.0的流动相,添加了10 mM乙酸铵和0.1%乙酸。
3. 1. 2. 质谱条件优化
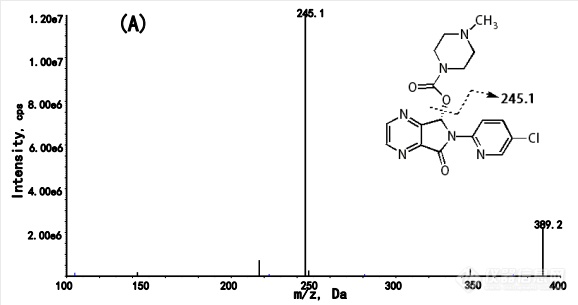
利用ESI-MS分析和MS参数优化正离子模式,提高了MRM测量对ESI源的响应。对于E-ZOP(图2),MRM的碎片化转变为m/z 389.2到245.1(图2),对于E-ZOP-d8(图2)为m/z 397.2到245.1,每个转变的停留时间为300 ms。每个化合物的碰撞能为22ev。分离电位保持在40 V。适用于所有分析物的源参数为15 psi的幕气、介质的碰撞气体(CAD)、温度为650℃、离子喷射电压为5000 V、离子源气体为60 psi。
3.1.3 样品处理方法优化
在样品制备的优化方面,与乙酸乙酯液-液萃取法相比,蛋白质沉淀法具有精度高、回收率高、操作简单等优点。样品制备采用蛋白质沉淀法。E-ZOP的定量限值可用于人血浆样品中药物动力学的定量分析。
最初,沉淀蛋白的溶剂是乙腈和甲醇,但这造成了E-ZOP含量的巨大损失,这可能是由于乙腈和甲醇不能有效地将分析员从蛋白质中解吸出来造成的。影响电荷态分布的因素包括溶剂pH值和药物溶解度。E-ZOP在水中的溶解度很低,在纯乙醇和正丙醇中的溶解度最低,这表明E-ZOP在醇中的溶解性很低,但在乙酸酯和酮中的溶解度较高[9]。因此,一种更易溶解的溶液二甲基亚砜与甲醇和甲酸按3:7:0.1的比例混合,成功地解决了这个问题。二甲基亚砜浓度高于此比值将显著降低仪器灵敏度。
3. 2. 方法验证
3. 2. 1. 选择性和专属性
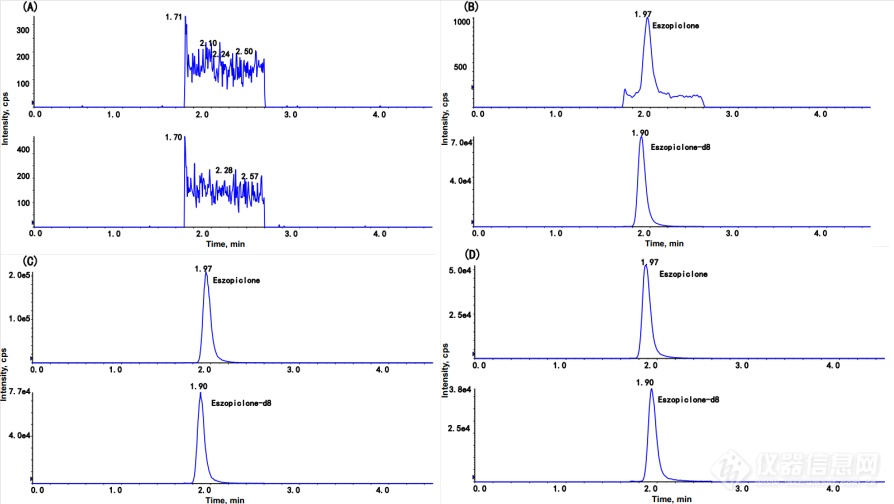
空白血浆、以2 ng/mL加入E-ZOP的空白血浆、以600 ng/mL加入E-ZOP的空白血浆以及口服E-ZOP后获得的临床血浆样本的代表性色谱图如图3所示。在所建立的色谱条件下,血浆中没有内源性干扰,表明该方法的选择性是可以接受的。
3. 2. 2. 线性和标准曲线
结果表明,在0.1~30 ng/mL血浆浓度范围内,E-ZOP的八点校准曲线呈线性关系,相关系数大于0.999。校准曲线的典型方程为:y=0.1x+0.00169,R2=0.9996,其中y代表E-ZOP-d8与IS的峰面积比,x代表E-ZOP的血浆浓度。以定量下限(LLOQ)作为校准曲线的最低浓度,其S/N比值均在10以上,精密度和准确度均可接受。
3. 2. 3. 精密度和准确度
LLOQ和QC样品中E-ZOP的精密度和准确度结果见表1。E-ZOP各样品水平的精密度(RSD)均小于9.99 %。对E-ZOP各样品水平的准确度在1.48 %到8.31 %之间。测定值均在可接受范围内。
3. 2. 4. 提取回收率和基质效应
在人空白血浆中,E-ZOP内标物均匀化的平均基质效应为101.2-106.0 %,而高溶血性的平均基质效应为99.9-101.4 %。在高脂血浆中,E-ZOP的基质效应为95.5-101.1 %。如表2所示,所有相对标准偏差值在0.75 %到9.17 %之间,这表明血浆基质的影响对于分析来说是可以忽略的。E-ZOP内标物均匀化后的平均提取回收率为96.7-103.8 %,不同浓度下E-ZOP的提取回收率结果准确、重现性好。
3. 2. 5. 稳定性
考察了E-ZOP在几种条件下的稳定性。E-ZOP在时间零点和室温下放置至少3h后,E-ZOP的CV%(7.35 %)的响应无显著差异(<15 %),表明E-ZOP在此条件下是稳定的。血浆样品在至少三个冷冻/解冻循环中的CV值(11.92 %)稳定。处理后的样品在自动进样器中稳定达24小时,在室温托盘中稳定达3小时,CV%值分别至少为6.88 %和7.35 %。血浆样品在-20℃下稳定至少4周,无明显损失(<8.43 %)。
3. 3. 药动学研究
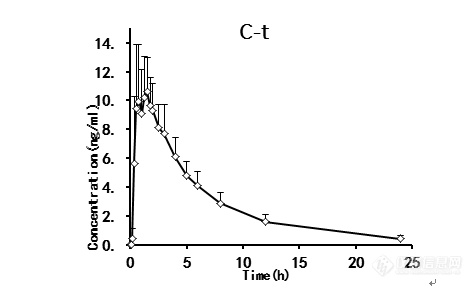
用完全有效的分析方法定量E-ZOP的人血浆PK浓度。口服E-ZOP后的平均血浆浓度-时间曲线如图4所示。药代动力学参数见表5。服用E-ZOP(1 mg)后,0.92±0.446h(Tmax)的最大观察血浆浓度(Cmax)为13.27±2.717 ng/mL。从时间零点到最后可测浓度(AUC0-t)的血药浓度-时间曲线下面积(AUC0-∞)分别为69.31±14.90 ng/mL•h和71.84±16.29 ng/mL•h。CLz/F为14.595±3.356l h,Vz/F为100.26±13.9l h,T1/2为4.90±0.819 h,所得数据表明E-ZOP可快速吸收,缓慢消除。与Hotha等人相比。报道了E-ZOP(3 mg)给药后的药代动力学参数,我们的药代动力学特性与以往文献相似[4]。
4. 结论
建立了一种简便、可靠的
LC-MS/MS法测定人血浆中E-ZOP的生物分析方法,具有选择性、灵敏、准确、准确等特点。与之前报道的样品制备方法相比,蛋白质沉淀法在灵敏度和操作简便性方面更为优越;该方法耗时更少,且具有高通量测试。用于方法验证,遵循FDA生物液分析规程。对12名健康志愿者进行单剂量口服1 mg E-ZOP后的药代动力学研究。计算了E-ZOP的PK数据,并与报告值进行了比较,证明了该方法在PK、生物等效性和生物利用度研究中的成功应用。
参考文献
[1] Liu, Shiyuan, et al. "Determination and correlation of solubility and thermodynamic properties of E-ZOP in pure and mixed solvents." Journal of Molecular Liquids 221 (2016): 1035-1044.
[2] Barnes, C. M., & Drake, C. L. (2015). Prioritizing sleep health: Public health policy recommendations. Perspectives on Psychological Science, 10, 733–737.
[3] Caldwell, J. A., Knapik, J. J., & Lieberman, H. R. (2017). Trends and factors associated with insomnia and sleep apnea in all United States military service members from 2005 to 2014. Journal of Sleep Research, 26, 665–670
[4] Derogatis, L. R., Lipman, R. S., Rickels, K., Uhlenhuth, E. H., & Covi, L.(1974). The Hopkins Symptom Checklist (HSCL): A self‐report symptom inventory. Behavioral Science, 19, 1–15.
[5] Dykema, J., Stevenson, J., Klein, L., Kim, Y., & Day, B. (2013). Effects of E-mailed versus mailed invitations and incentives on response rates, data quality, and costs in a web survey of university faculty. Social Science Computer Review, 31, 359–370.
[6] Sivertsen B, Vedaa Ø, Harvey A G, et al. Sleep patterns and insomnia in young adults: A national survey of Norwegian university students[J]. Journal of Sleep Research, 2018: e12790.
[7] El-Yazbi AF, Youssef R M. An eco-friendly HPTLC method for assay of E-ZOP in pharmaceutical preparation: investigation of its water-induced degradation kinetics [J]. Analytical Methods, 2015, 7(18): 7590-7595.
[8] Men
gcai Z, Songwei P. Bioequivanlence of Zopiclone Tablet in Healthy Volunteers [J]. China Pharmacist, 2010, 3: 011.
[9] Marin, Stephanie J., et al. "Sensitive UPLC–MS-MS assay for 21 benzodiazepine drugs and metabolites, zolpidem and zopiclone in serum or plasma." Journal of analytical toxicology 36.7 (2012): 472-476.
[10] Olsen R W. GABA-benzodiazepine-barbiturate receptor interactions [J]. Journal of neurochemistry, 1981, 37(1): 1-13.
[11] Najib J. E-ZOP, a nonbenzodiazepine sedative-hypnotic agent for the treatment of transient and chronic insomnia [J]. Clinical therapeutics, 2006, 28(4): 491-516.
[12] Kumar R N, Rao G N, Naidu P Y. Stability Indicating RP-LC Method for Determination of E-ZOP in Bulk and Pharmaceutical Dosage Forms [J]. Asian journal of research in chemistry, 2010, 3(2): 374-379.
[13] CHEN Q, LIU Y, REN X, et al. Relative Bioavailability and Bioequivalence of Zopiclone Tablets in Healthy Volunteers [J]. Herald of Medicine, 2012, 11: 005.
[14] Greenblatt D J, Zammit G K. Pharmacokinetic evaluation of E-ZOP: clinical and therapeutic implications[J]. Expert opinion on drug metabolism & toxicology, 2012, 8(12): 1609-1618.
[15] Meng, Min, et al. "Fast chiral chromatographic method development and validation for the quantitation of E-ZOP in human plasma using LC/MS/MS." Journal of pharmaceutical and biomedical analysis 53.4 (2010): 973-982.
[16] Paw B, Misztal G. Chromatographic analysis (TLC) of zopiclone and benzodiazepines [J]. JPC-JOURNAL OF PLANAR CHROMATOGRAPHY-MODERN TLC, 2000, 13(3): 195-198.
[17] Sharma N, Rao S S, Kumar N D A, et al. A Novel Validated Ultra-Performance Liquid Chromatography Method for Separation of E-ZOP Impurities and its Degradants in Drug Products [J]. Journal of AOAC International, 2013, 96(5): 981-986.
[18] Dhaneshwar S R, Vidhya K B. Development of a Validated Stability-Indicating HPLC assay method for E-ZOP [J]. International Journal of ChemTech Research, 2011, 3(2): 680-689.
[19] Tracqui A, Kintz P, Manqin P. High‐performance liquid chromatographic assay with diode‐array detection for toxicological screening of zopiclone, zolpidem, suriclone and alpidem in human plasma. Journal of Chromatography: Biomedical Applications 1993; 616: 95–103.
[20] Paw W and Misztal G. Determination of zopiclone in tablets by HPLC and UV spectrophotometry. Journal of Pharmaceutical and Biomedical Analysis 2000; 23: 819–823.
[21] Foster RT, Caille G, Ngoc AH, Lemko CH, Kherani R and Pasutto FM. Stereospecific high‐performance liquid chromatographic assay of zopiclone in human plasma. Journal of Chromatography B 1994; 658: 161–166.
[22] Gebauer MG and Alderman CP. Validation of a high‐performance liquid chromatographic method for the enantiospecific quantitation of zopiclone in plasma. Biomedical Chromatography 2002; 16: 241–246.
[23] Van Bocxlaer J, Meyer E, Clauwaert K, Lambert W, Piette M and DeLeenheer A. Analysis of zopiclone (Imovane) in postmortem specimens by
gc–MS and HPLC with diode‐array detection. Journal of Analytical Toxicology 1996; 20:52–54.
[24] Gaillard Y, Gay‐Montchamp JP and Ollagnier O. Gas chromatographic determination of zopiclone in plasma after solid‐phase extraction. Journal of Chromatography 1993; 619: 310–314.
[25] Chennaiah M, Veeraiah T, Venkateshwarlu G. Spectrophotometric determination of E-ZOP in pure and pharmaceutical forms [J]. Journal of the Chilean Chemical Society, 2012, 57(4): 1460-1463.
[26] Oiestad L, Johansen U and Christophersen AS. Drug screening of preserved oral fluid by liquid chromatography–tandem mass spectrometry. Clinical Chemistry 2007; 53: 300–309.
[27] Ouintela O, Sauvage FL, Charvier F, Gaulier JM, Lachatre G and Marquet P. Liquid Chromatography–tandem mass spectrometry from detection of low concentration of 21 benzodiazepines, metabolites, and analogs in urine: method with forensic application. Clinical Chemistry 2006; 52: 1346–1355.
[28] Laloup M, Ramirez Fernanadez M, De Boeck G, Wood M, Maes V and Samyn N. Validation of a liquid chromatography–tandem mass spectrometry method for the simultaneous determination of 26 benzodiazepines and metabolites, zolpidem and zopiclone, in blood, urine and hair. Journal of Analytical Toxicology 2005; 29: 616–626.
[29] Kratzsch C, Tenberken O, Peters FT, Weber AA, Kraemer T and Maurer HH. Screening, library‐assisted identification and validated quantification of 23 benzodiazepines, flumazenil, zaleplone, zolpidem and zopiclone in plasma by liquid chromatography/mass spectrometry with atmospheric pressure chemical ionization. Journal of Mass Spectrometry 2004; 39: 856–872.
[30] Hotha K K, Vijaya Bharathi D, Jagadeesh B, et al. A rapid
LC-MS/MS method for quantitation of E-ZOP in human plasma: application to a human pharmacokinetic study [J]. Biomedical Chromatography, 2012, 26(2): 225-231.
[31] Ravi D, Rao J S, Rajalakshmi D, et al. Development and validation of an RP-HPLC Method for the determination of the E-ZOP in tablet dosage forms [J]. Inter. j. of res in pharm and Chem, 2014, 4: 119-130.
[32] Shaikh K, Patil A, Gite S. Stability-Indicating LC–UV Method for the Determination of E-ZOP and Degradation Impurities in Tablet Dosage Form [J]. Journal of chromatographic science, 2013, 52(4): 293-297.
[33] Anandakumar K, Kumaraswamy G, Ayyappan T, et al. Quantitative Estimation of E-ZOP in Bulk and in Formulation by Simple UV and Difference Spectroscopic Methods[J]. Research Journal of Pharmacy and Technology, 2010, 3(1): 202-205.
[34] Liu S, Xu S, Du S, et al. Determination and correlation of solubility and thermodynamic properties of E-ZOP in pure and mixed solvents[J]. Journal of Molecular Liquids, 2016, 221: 1035-1044.
[35] Salama N, Zaazaa H, Abd El Halim L, et al. Thin-layer chromatographic enantioseparation of ofloxacin and zopiclone using hydroxy-propyl-beta-cyclodextrin as chiral selector and thermodynamic studies of complexation[J]. JPC-Journal of Planar Chromatography-Modern TLC, 2014, 27(3): 166-173.
[36] Bioanalytical method validation, biopharmaceutics coordinating committee in the Center for Drug Evaluation and Research (CDER) in cooperation with the Center for Veterinary Medicine (CVM) at the Food and Drug Administration. Guidance for Industry, (2001), Accessed date: May 2018.
表格
表1人血浆中E-ZOP的精密度和准确度。Concentration(ng/mL) | intra-day(n=6) | inter-day(n=18) |
Precision(RSD%) | Accuracy(%) | Precision(RSD%) | Accuracy(%) |
0.1 | 4.8 | 109.2 | 8.7 | 104.2 |
0.25 | 2.8 | 108.2 | 6.7 | 104.5 |
2.5 | 1.6 | 96.7 | 3.6 | 98.4 |
25 | 2.3 | 96.8 | 2.6 | 96.6 |
表2 E-ZOP的提取回收率和基质效应(n=6)。Compound | plasma types | Sample Conc.(ng/mL) | Matrix effect | Extraction recovery |
Mean±SD (%) | RSD% | Mean±SD (%) | RSD% |
E-ZOP | blank plasma | 0.25 | 104.2±2.6 | 2.5 | 93.6±6.0 | 6.5 |
2.5 | 101.4±1.8 | 1.8 | 93.4±1.8 | 1.9 |
25 | 94.4±7.0 | 7.4 | 108.3±2.9 | 2.7 |
high hemolysis | 0.25 | 100.5±7.3 | 7.3 | | |
2.5 | 100.9±1.8 | 1.7 | | |
25 | 101.0±2.0 | 2.0 | | |
high fat plasma | 0.25 | 96.3±3.7 | 3.9 | | |
2.5 | 100.1±1.4 | 1.4 | | |
25 | 101.7±1.5 | 1.5 | | |
表3 E-ZOP的样品稳定性(n=6,以平均值±R.E.%)表示。Stability test condition | Norminal concentration added(ng/mL) |
E-ZOP in plasma |
0.25 | 2.5 | 25 |
Long-term stability | | | |
7 days at -20℃ | 0.27±0.01 | 2.56±0.05 | 24.75±0.45 |
45 days at -20℃ | 0.27±0.02 | 2.48±0.1 | 24.6±0.45 |
Short-term stability | | | |
Bench-top-3h | 0.23±0.02 | 2.31±0.03 | 22.10±0.38 |
Autosampler-24h | 0.26±0.02 | 2.45±0.03 | 24.25±0.53 |
Freeze and thaw stability 3 cycles at -20 ℃ | 0.27±0.01 | 2.55±0.06 | 26.21±1.36 |
表4 E-ZOP溶液稳定性(n=6)。Sample Conc.(ng/mL) | E-ZOP-0day | E-ZOP-7day | E-ZOP-45day |
Mean±SD (%) | RSD% | Mean±SD (%) | RSD% | Mean±SD (%) | RSD% |
0.25 | 0.02±0.00 | 12.33 | 0.03±0.00 | 9.16 | 0.02±0.00 | 5.19 |
25 | 2.17±0.03 | 1.4 | 2.19±0.08 | 3.46 | 2.19±0.03 | 1.57 |
表5口服E-ZOP后的非房室药代动力学参数(平均值±标准差,n=12)。
Oral admin (mg) | Parameters | |
T1/2 | Cmax | Tmax | AUC(0-t) | AUC(0-∞) | CLz/F | Vz/F |
(h) | (ng/mL) | (h) | (ng/mL·h) | (ng/mL·h) | (L/h) | (L) |
1 | 4.90±0.819 | 13.27±2.717 | 0.92±0.446 | 69.31±14.90 | 71.84±16.29 | 14.595±3.356 | 100.26 ± 13.9 |
亮点•采用一种新的LC-MS/MS方法,通过简化的蛋白质沉淀进行单步预处理。•高通量和每次进样4.5分钟。•开发并验证了特定且灵敏的LC-MS/MS方法。•根据美国食品和药物管理局的规定,采用可接受的标准对所建立的方法进行了验证。•已验证的方法用于测定人血浆中的E-ZOP。•该方法可用于临床试验中E-ZOP的定量和监测。•选定的同位素内标可有效减少基体效应和误差。![]()
图 1
![]()
![]()
图 2
![]()
图 3
![]()
图 4
图1. (A)E-ZOP和(B)E-ZOP-d8的结构(内部标准)
图2. A(E-ZOP)和B(E-ZOP-d8)的[M+H]+全扫描产物离子质谱。
图3. 人血浆中E-ZOP和E-ZOP-d8的代表性MRM色谱图。(A) 空白血浆样品;(B)加入E-ZOP(2 ng/mL)和E-ZOP-d8的空白血浆;(C)加入E-ZOP(600 ng/mL)和E-ZOP-d8的空白血浆;(D)人体口服E-ZOP(1 mg)0.5h后的血浆样本。
图4. 口服1 mg/kg(n=12)后E-ZOP的平均药代动力学曲线