三、方法学考察: 现已***片为例,进行方法学研究。在***片中选择其君药中所含的A成分作为指标性成分进行含量测定的方法学研究。
3.1 检测波长的选择:取A对照品溶液,在200~700nm波长进行光谱扫描,发现光谱图在530nm波长处有最大吸收,故测定波长选定为530nm。
说明:一般选择待测样品化合物吸收度最大,即吸收曲线最高点为测定波长。化合物的最大吸收峰λmax或该化合物经显色后的最大吸收峰,通过分光光度计进行扫描后确定或通过二极管阵列检测来确定,并与该化合物文献值相比较应一致。在最大吸收峰处测定时灵敏度高,误差小。因此一般情况下选择最大吸收波长作为检测波长。
3.2 样品提取方法、溶剂考察:
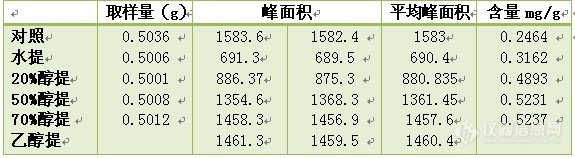
取***片0.5克,精密加入不同溶剂25ml进行超声提取30分钟,进行含量测定,计算目标成分的含量。结果见下表:
![]()
以上结果表明,70%醇提取和乙醇提取含量最高,而二者含量又无明显差异,故确定提取溶剂为70%乙醇。
考察方法:根据指标性成分的性质选用易提取的几种溶剂进行提取对比试验,选择提取率高的一种溶剂作为提取溶剂。
3.3 提取时间考察:取***片0.5克,精密加入70%乙醇进行超声提取5、10、15、20、30分钟,进行含量测定,计算目标成分的含量。结果见下表:
![]()
以上结果表明,超声处理15分钟,含量不再增加,为了保证提取完全,故确定超声处理时间为20分钟。
考察目的:主要是选择把目标成分完全提取出来,所需的最短的时间,以便制定合理的分析方法。
3.4 标准品纯度考察:对照品纯度要求达到98%以上。对照品纯度一般都能够达到要求。
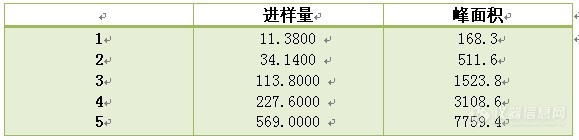
3.5 标准曲线制作:配制不同浓度的A对照品溶液,考察线进样量与峰面积的性关系、线性范围、相关系数等。
![]()
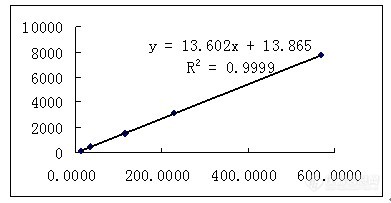
以进样量为横坐标峰面积为纵坐标做标准曲线:
![]()
以上结果表明,在0.0114~0.0569μg范围内,本品峰面积值与进样量有良好的线性关系。
说明:做标准曲线的目主要是(1)确定样品进样量与峰面积是否呈线性关系;(2)、确定线性范围,即适用的样品进样量的确定;(3)、标准曲线是否通过原点,通过原点时,选用一种浓度的标准品称一点法,进行含量测定;不通过原点时,选用两种浓度的标准品(称两点法)进行含量测定。
3.6 阴性试验:要求样品溶液在与对照品
液相同位置有相同保留时间的色谱峰,而阴性则无,主要是考察方法的专属性。常用阴阳对照法,即含以被测成分的药材样品与除去该成分的药材的样品作对照,可考察被测成分的位置是否与干扰组分重迭,以确证测定指标是否仅为被测成分的响应,防止假阳性的误判。
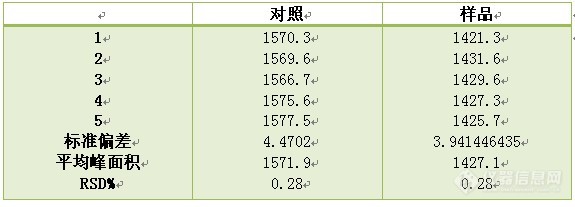
3.7 稳定性试验:考察不同时间点是否对测定方法和测定结果有影响,用同一被测样品的供试液在不同间隔时间用同一测定方法所得到的测定结果。一般考察36小时,计算RSD%不得大于3%。对照和样品均要做.
稳定性试验
![]()
试验结果表明,样品供试液和对照品溶液在24小时内稳定性良好
3.8 精密度试验:是指用相同方法对同一样品溶液进行多次测定,考察各测定值彼此接近的程度。具体如下:取同一样品,连续测定五次,相对标准偏差RSD%不得大于3%。对照和样品均要做 ,具体如下:
精密度试验
![]()
试验表明,仪器精密度良好。
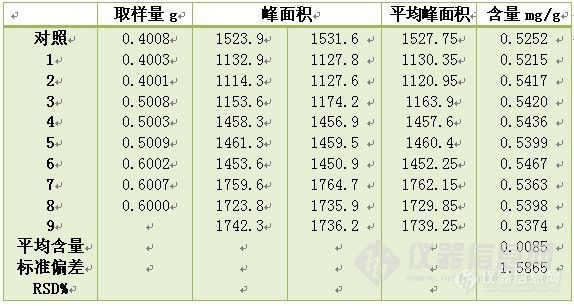
3.9 重复性试验:是指在同一条件下对同一批样品,从样品供试品液制备始,制备多份供试品溶液。每份供试品液再分别进行测定,测定所得到的数据进行统计学处理,计算其含量的平均值和相对标准偏差(RSD%)。具体如下:同一批号样品,分别取低、中、高三个样品量,每个样品量3份,按样品测定方法操作,相对标准偏差RSD%不得大于3%。
![]()
3.10 加样回收试验:一般回收率要求在95~105%。
![]()
![]()
试验结果表明:回收率在95%~100%之间,加样回收良好
详解:加样回收试验即于已知被测成分含量的成药中再精密加入一定量的被测成分纯品,依法测定。用实测值与原样品中含测成分之差,除以加入纯品量计算回收率。此法不用制备空白对照,模拟真实性好。
注意事项:1、纯品的加人量与取样量中被测成分之和必须在标淮曲线线性关系范围之内;2、外加纯品的量要适当,过小则引起较大的相对误差,过大则干扰成分相对减少,真实性差。3、一般加入量与所取样品含量之比控制在1:1左右。4、做加样试验时,有人将对照品加至制备好的供试品溶液中,这是不对的,这样不能考察提取、纯化过程中被测成分是否损失,不能代表含量测定方法的回收率。因此要在称样开始时就加入对照品!
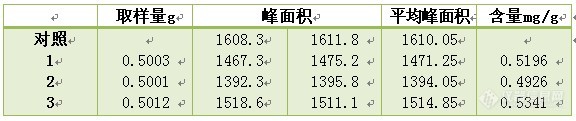
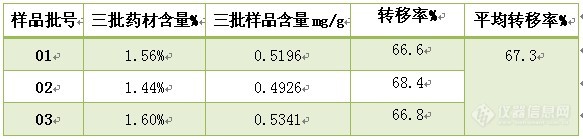
3.11 含量限度的制定:申报临床,必须根据原料来源不同的三批以上样品测定结果,确定含量范围
三批样品含量
![]()
每克样品中含某药材量0.5克
计算转移率 ![]()
制定含量限度:由上表可知A成分的平均转移率为67.3% ,而每克样品中含某药材量0.5克,中国药典2005版某药材项下规定的A成分不低于1.0 %,因此确定,每克样品中A成分的含量限度为:0.5×1.0%×67.3%=0.3365mg/g。常用的含量表示方法有:%、mg/片、"g/丸、mg/m1(液体制剂)等。
![]()