获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
风之彩回复于2008/11/20
倒峰产生的原因:
1 信号线极性接反了或是参数设置成倒峰的;
2 走梯度走出来的;
3 溶剂峰
4 氘灯老化,流通管道或池不通畅;
5 样品和流动相中含有小颗粒,气泡等;
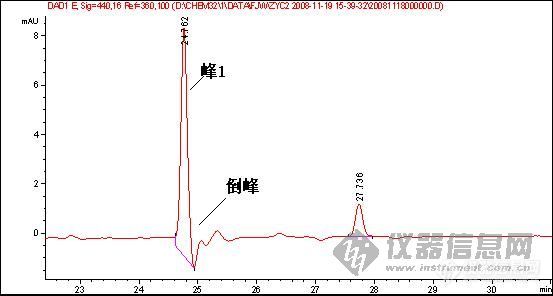
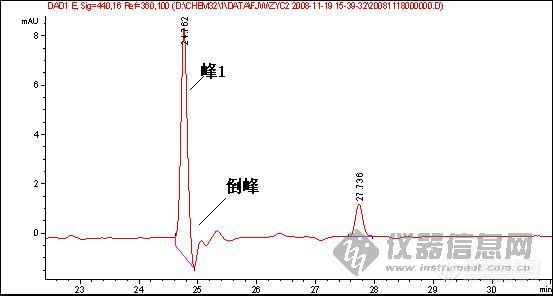
6 参比波长设置的不合适,如楼主的图,参比波长选择360nm,如果在出峰位置的某化合物在360nm左右的某一范围内有吸收,那么就可能出现倒峰
前面两张图的倒峰不是很明显,特别是第二个倒峰像是基线波动,样品峰后面的倒峰是不是样品溶剂的关系?换个溶剂看看?或是这个地方有干扰?调整保留时间看看?
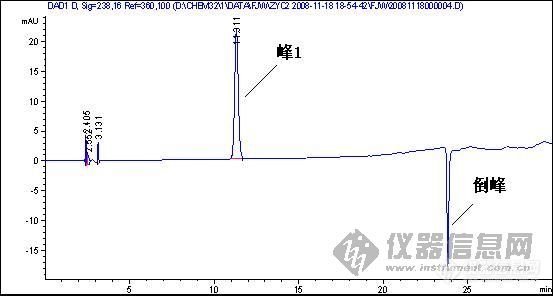
第3张图可能都是第6种原因导致,建议把参比波长重新设置,或是干脆把参比波长关掉看看,可能就会好了。
tanghongmin回复于2008/11/19
我基本赞成1、2楼的hyban的观点,但是对于已经25min出峰后的倒峰,应该不会是溶剂峰,溶剂峰出来比较早,一般在5min前,所以用流动相来配比定容供试液不会有作用
倒峰产生的原因我认为主要有两个,就如1、2楼的hyban说的检测器问题,光源不稳定,背景噪音大,如果我没有说错的话,你用的这个检测器是Agilent的紫外或是DAD(仪器用的比较多的板油做仪器耐用性的时候就能对比出来);另外一方面是当流动相极性比例发生大幅度改变时,泵后流动相由于死体积大或者泵来不及调整导致流动相混合不均匀,或者气泡没有完全除干净,温度未恒温等原因都会产生倒峰
ygx回复于2008/11/19
1.倒峰的出现,无怪乎有一下几个原因:试样的溶剂峰;试样中的杂质峰(且含量较大);流动相中有气泡;检测器性能不稳等。
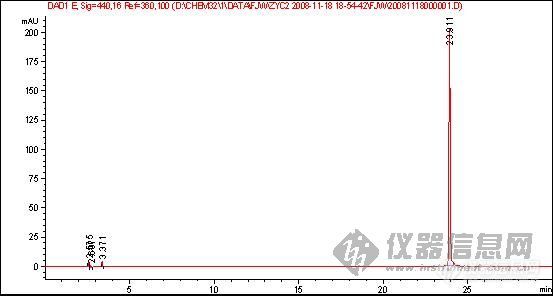
2.第三幅图的倒峰应该不是溶剂峰。对于峰漂移的问题,对照品浓度和样品浓度应尽可能的接近,另外两者稀释的溶剂也应一致。
3.对于第三幅图,应该对峰1的定量不会带来多大影响的。
对于第一幅图,应该在流动相、检测器方面着手改进或完善:若是气泡问题,就对流动相彻底脱气;若是杂质问题,就改变流动相组成或配比,或者由恒流变梯度,或变换检测器波长等;若是溶剂的问题,就更换溶剂或用流动相稀释;若是检测器的性能问题,就降低检测器的灵敏度或更换光源等。
吹不动的浮云回复于2008/11/21
鉴于你的这个试验,在25分钟总会出一个倒峰,只是你的峰1的出峰时间在变,如果可以确定在25分钟是一个成分,我建议更换低吸收的流动相,定量需要重现,现在看来你做的样品不能够保证样品的保留时间重现,一是建议多稳定一段时间,二是,保证进样条件和进样量重现,三,保证样品不被污染和溶液成分的改变!(乙腈和水的吸收较小)
原文由 03yx2 发表:大家图谱来找茬(第三季) ◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆
请您来分析:
1.产生倒峰的原因有哪些?
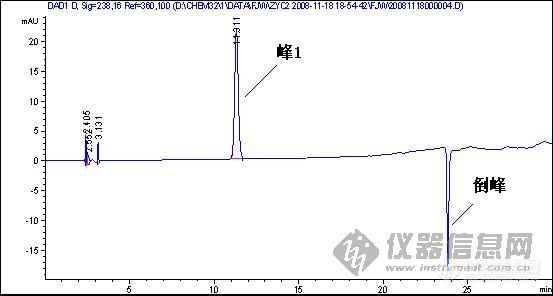
2.图谱中一个重要问题是倒峰,一个是离峰1很近的倒峰,一个是离峰1比较远,请问有什么区别?针对峰的漂移你有什么好的建议嘛?
3.如果要对图中的峰1进行含量测定,你觉得应该怎样改善条件才可以更好的进行定量分析.
〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓〓
图谱如下:
1、样品峰1:
◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇
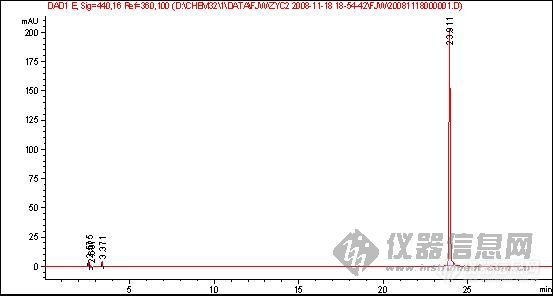
2、该物质的对照品图谱:
◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇◇
3、另一类倒峰:
【附录】凡参与活动者,必有重奖