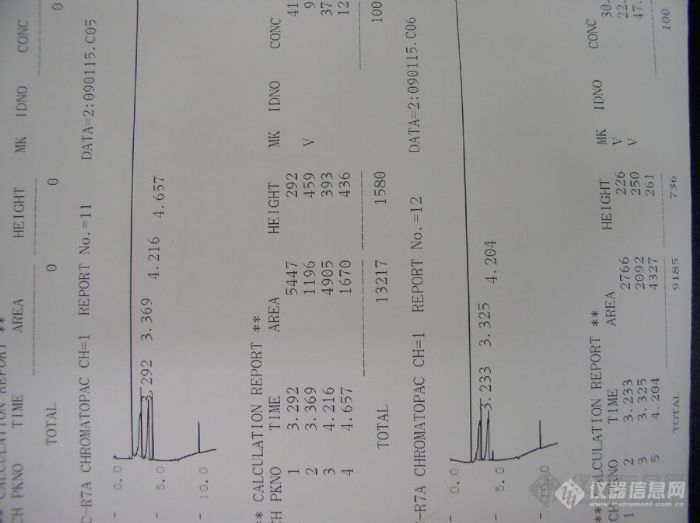
用毛细管柱测定法莫替丁中的甲醇、乙醇和NN二甲基甲酰铵中的有机残留,氮气做载气,FID检测器;进样口温度40,保留3分钟,25度每分的升温速率到150度,保留5分钟。可是进针后有时候出现一个类似的溶剂峰,有时候又没有,而且同一个条件下进一个配制好的标准样品,出峰形状也不同,主一是后面的三个被测的有机溶剂峰形太难看,请大家帮忙分析一下,急死我了。传上来的图分别为:1图为不进样空走图,2、3图为公司自制超纯水(结果不同),4、5图为卖来的注射用水(也不一样),6、7图为自己配制的甲醇、乙醇、NN二甲基甲酰胺标准液(结果也不同)。
先声明:这种组分分析我没做过,可能会有说不到位的情况,莫见怪.
1,进样问题,是手动进,还是自动进,时有时没有的溶剂峰,是否考虑检查一下进样垫?
2,数据结果不同的问题.看图说话,好像是积分仪谱图,对于结果不同,针对你的谱图峰来看,如果峰形好,用峰高定量,峰形不好,只能用峰面积啦;
3,谱图不好,应该与你的柱子和操作条件有很大关系,能否尝试一下,用恒温进样模式.而且我对3分钟才程升很疑惑,为什么,这不是与那几个样对着干吗?往后拖2分钟即恒温5分钟再程升如何?我觉得你程升的目的是针对最后一个峰的,如果是针对前几个,程升是否晚了点,而且幅度这么大,柱压干扰因素肯定明显.
以上仅是个人判断,决定权在你!