顶空气相色谱系列讲座(106)-吉非罗齐中挥发性酯类溶剂 顶空进样-毛细管
气相色谱法检测吉非罗齐中残留溶剂
摘要 目的:建立顶空进样-毛细管
气相色谱法,以乙酸异丁酯为内标,检测吉非罗齐中甲酸乙酯、四氢呋喃与异丁酸异丁酯。
方法:采用键合聚乙二醇色谱柱(DB -WAX,0.53 mm×30 m×1μm);柱温采取程序升温的方法;顶空进样;载气为氮气。
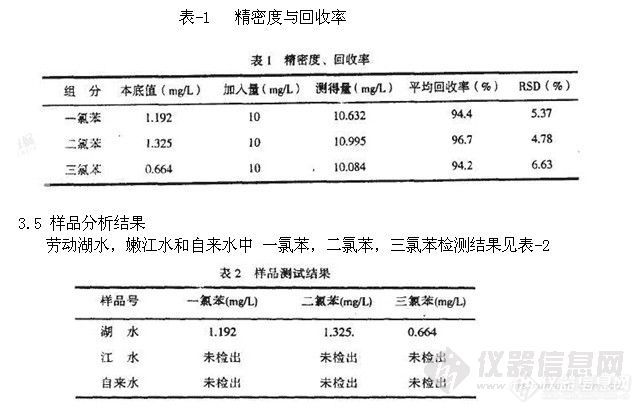
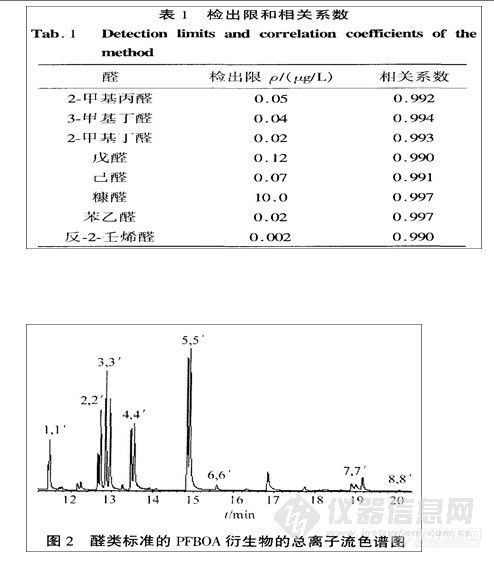
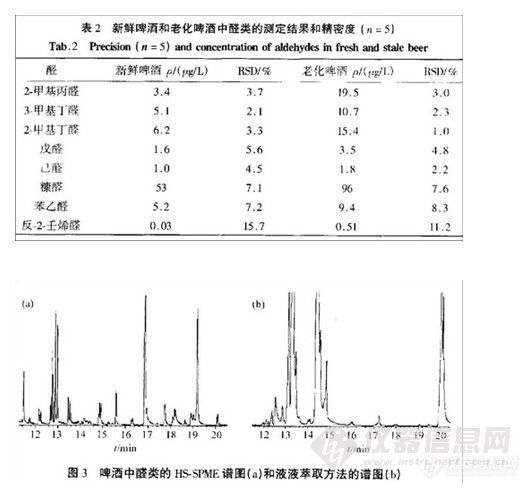
结果:在本色谱条件下,测得各组分的线性关系良好(相关系数均在0.9997以上);平均回收率分别为100.6%,99.4%,99.8%;最小检出浓度分别为0.194,0.195,0.509
μg·mLˉ1
结论:本法灵敏、准确,适用于吉非罗齐中甲酸乙酯、四氢呋喃与异丁酸异丁酯残留溶剂的检测。
本文由湖州市药品检验所的蒋国强同志撰写的实验结果,与前面介绍的分析残留溶剂品种有所不同,特选出并具体介绍如下。
前言
吉非罗齐是一种降血脂药,化学名为2,2-二甲基-5-(2,5-二甲苯基氧基)-戊酸[l]。本品在合成过程中用到了多种有机溶剂,文献报道[1]采用2个系统同时检测,但样品处理复杂。为了有效地控制产品的质量和用药安全,本文建立顶空进样-
气相色谱法,检测吉非罗齐中溶剂的残留量,方法简便、灵敏、准确。
实验
1 仪器与试药
HP-6890
气相色谱仪;顶空进样器Agilent 7694E Headspace Sampler;FID检测器及分析仪;甲酸乙酯、四氢呋喃、异丁酸异丁酯与乙酸异丁酯(内标)对照品均为分析纯,N,N-二甲基甲酰胺为色谱纯;样品为吉非罗齐原料药(浙江精进医药化工有限公司,批号030224,030225,030226)。
2 方法与结果
2.1 色谱条件
色谱柱:DB-WAX键合聚乙二醇色谱柱(0.53 mm×30 m×1 μm);柱温采用程序升温:初始温度40℃,保持3 min,5℃·minˉ1升温至60℃,再10℃·minˉ1升温至100℃,最后以50℃·minˉ1升温至200℃,保持10 min;顶空进样,定量管温度:100℃,传输管温度:110℃;进样口温度:200℃;检测器温度:250℃;载气为氮气,流速2.0 mL·minˉ1分流比为1:1。
2.2 试验法
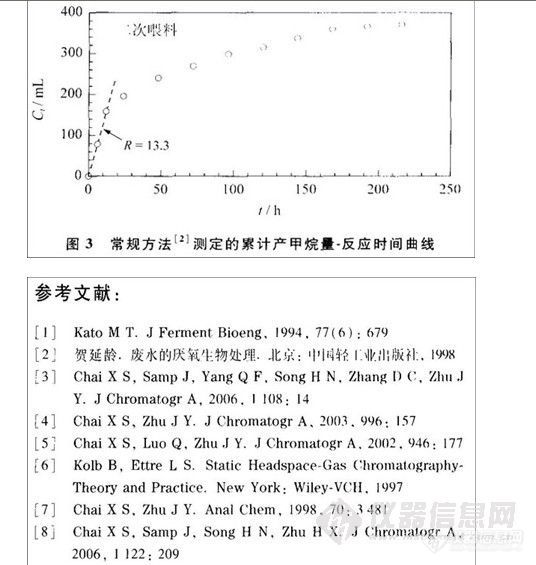
取乙酸异丁酯适量,加N,N-二甲基甲酰胺稀释、混匀,制成约2 mg·minˉ1的溶液,作为内标储备液;精密量取内标储备液5 rnL,置100mL量瓶中,加N,N-二甲基甲酰胺稀释至刻度,摇匀,作为内标溶液;取甲酸乙酯、四氢呋喃与异丁酸异丁酯各100 mg,精密称定,置同- 50 mL量瓶中,加N,N-二甲基甲酰胺稀释至刻度,摇匀,作为对照品储备液;精密量取对照品储备液与内标储备液各5 mL,置100 mL量瓶中,加N,N-二甲基甲酰胺稀释至刻度,摇匀,精密量取5 mL,置顶空取样瓶中,作为对照品溶液;取本品约100 mg,精密称定,置顶空取样瓶中,精密加人内标溶液5 mL,振摇使溶解,作为供试品溶液。取对照品溶液与供试品溶液,于90℃加热30 min,照中国药典2005年版二部残留溶剂测定法[3],在本文色谱条件下检测。甲酸乙酯峰与四氢呋喃峰的分离度应不低于2.0,异丁酸异丁酯与相邻峰的分离度应不低于7.0,按内标法以峰面积计算甲酸乙酯、四氢呋喃和异丁酸异丁酯的含量。色谱图见图1。
![]()
图-1 毛细管
气相色谱图
1. 甲酸乙酯 2. 四氢呋喃 3. 内标乙酸异丁酯 4. 异丁酸异丁酯 5. N,N-二甲基甲酰胺
2.3 线性试验
精密量取上述对照品储备液10mL,置50 mL量瓶中,加N,N-二甲基甲酰胺稀释至刻度,摇匀,分别精密量取1,2,3,4,5,6,7 mL,置20mL量瓶中,精密加人内标储备液1 mL,用N,N一二甲基甲酰胺稀释至刻度,摇匀,精密量取5 mL,分别置顶空取样瓶中,照上述色谱条件进样,测得峰面积。以各组分的浓度为横坐标,各组分峰面积与内标峰面积之比为纵坐标进行线性回归,结果甲酸乙酯、四氢呋喃、异丁酸异丁酯回归方程分别为:
R=-0.03445+23.73C r=0.9997
R= -0.02832+37.24C r=0.9998
R = 0.01258+5.400C r=0,9998
线性范围分别为0.019~0.14,0.02~0.14,0.02~0.14μg·mLˉ1。
2.4 精密度试验
取上述对照品溶液,照上述色谱条件,连续进样6次,计算各成分的校正因子,结果甲酸乙酯、四氢呋喃、异丁酸异丁酯平均校正因子(n=6)分别为0.044,0.027,0.17;RSD均为0.2%。
2.5 最低检测限度试验
取对照品储备液,分别用N,N-二甲基甲酰胺稀释制成每1 mL含甲酸乙酯1.94,0.97,0.485,0.194,0.097 μg;含四氢呋喃1.95,0.98,0.488,0.195,0.098 μg;含#]^沔菠5苹丁酯2.04,1.02,0.509,0.204,0.102 μg共5种不同浓度的混合溶液,照上述色谱条仵分别进样。甲酸乙酯、四氢呋喃、异丁酸异丁酯的最低检测限分别为0.194,0.195,0.509μg·mLˉ1
2.6 回收率试验
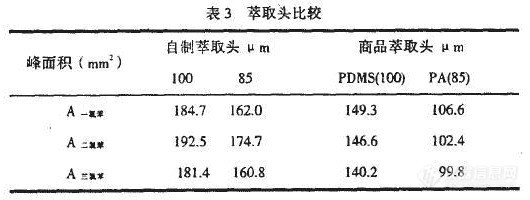
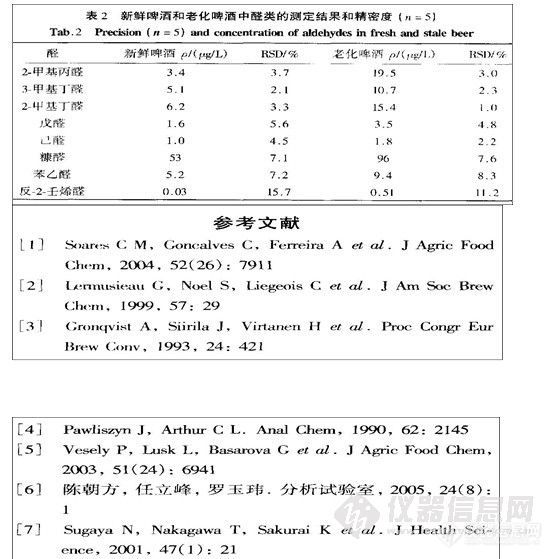
取批号为030225的样品约100mg,精密称定,精密加入对照品溶液和内标储备液各5 rnL,振摇使溶解,按“2.2”项下的方法进行操作,结果甲酸乙酯、四氢呋喃、异丁酸异丁酯平均回收率(n=2)分别为100.6%,99.4%,99.8%;偏差分别为2.4%,1.5%,1.9%。
2,7 样品测定
按“2.2”项下的方法对批号为030225,030226,030224的吉非罗齐原料药样品中甲酸乙酯、四氢呋喃、异丁酸异丁酯进行检测,结果检测到030225号样品中有甲酸乙酯(含量为0.002%)。
3 讨论
3.1 溶剂的选择
本品在水中不溶,在甲醇中溶解,在N,N-二甲基甲酰胺和氯仿中易溶。故选择溶解性好且对待测各组分均无干扰的N,N-二甲基甲酰胺为溶剂。
3.2 色谱柱的选择
本品控制的甲酸乙酯、四氢呋喃、异丁酸异丁酯3种有机溶剂的沸点和极性均有不同程度的差异。且均有一定的极性,根据相似相溶的原则,选择极性柱DB-WAX,3种有机溶剂、内标及溶剂均能达到较好的分离效果。
3.3 柱温及顶空条件的选择
本试验采用程序升温的方法,测定时柱温先在40℃保持3 min,5℃·minˉ1升温至60℃,再10℃·minˉ1升温至100℃,是为了使各待测组分更好地分离;最后以50℃·minˉ1升温至200℃,保持10 min,可以较快除尽溶剂,避免影响以后样品检测。采用溶液直接进样法测定时,主药干扰分析,同时色谱柱污染严重,而采用顶空进样则色谱柱污染少,色谱系统稳定;同时,由于3种有机溶剂的沸点均较高,顶空进样时顶空瓶于90℃加热30 min,定量管温度设置为100℃、传输管温度为110℃。
参考文献
1 CHEN Xue -fan(陈雪帆),JIA Eel(贾飞)Determination residual organic volatile solvents in gemfibrozil by capillary gas chromatography(
气相色谱法测定吉非罗齐中有机溶剂残留量)Drug Identification(药物鉴定),2004,13(8):29
2 CHENO Neng- lin(程能林) Solvents Reference(溶剂手册) Bei-Jing(北京):Chemical Industry Press(化学工业出版社),2002
3 ChP(中国药典)2005 Vol Ⅱ(二部):Appendix(附录)Ⅷ P