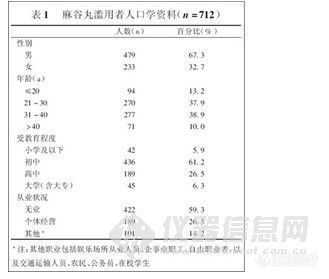
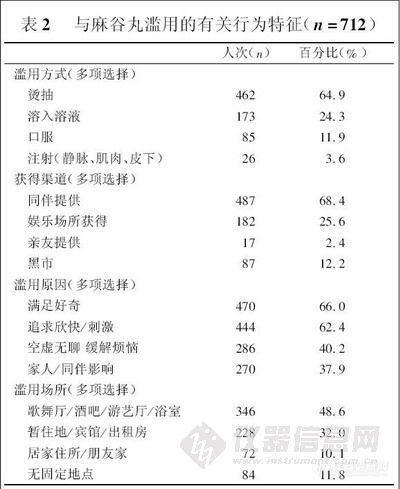
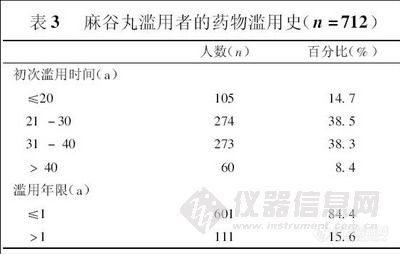
兴奋剂类分析方法系列讲座(5)-尿中氯胺酮及其代谢物
应用gc-双质谱法快速测定尿液中氯胺酮及其代谢物 摘 要 目的:应用串联质谱(CI离子源)法进行尿中氯胺酮及其代谢物的快速测定。
方法:取尿液2~3 mL,加入固体碳酸钠少量和丙酸酐80 μL,超声5 min,加入醋酸甲酯2 mL,超声萃取l0 min后离心(4000 r·minˉ1)3 min,取上层有机相转入事先加入无水硫酸钠的样品瓶(4 mL容量)中,供
gc - MS/MS测定。
结果:本方法不仅适用于尿中氯胺酮原药的检验,同时满足尿中去甲氯胺酮、脱氢去甲氯胺酮的检验,回收率达90%以上,最低检出浓度为:5×10-8 g·Lˉ1;
gc- MS/MS的最低检出限为0.05 ng。
结论:本方法可满足确证48 h以内是否吸食过氯胺酮。
前 言
近年来,因为吸食少量氯胺酮一般情况下不易成瘾,麻醉剌激效果又好,并且持有或贩卖达到一定数量才能构成犯罪,所以在娱乐场所吸食氯胺酮的案件呈快速上升趋势。以前的检验方法多以检验原形物氯胺酮为基准,目前必须重视体内的代谢问题,氯胺酮在人体内代谢分解快,超过l0 h的尿液中基本就检不出氯胺酮,所以给处理这类吸毒案件带来了难度。
氯胺酮进入人体后,主要经肝脏生物转化,并从尿液中排出体外。尿液中原形药仅占用药量的5%,其余则为代谢物,如去甲氯胺酮、脱氢去甲氯胺酮及羟化代谢物的葡萄糖醛酸苷结合物[1,2] 。为寻找一种快速、灵敏、可靠的检验方法,通过实验研究和临案应用,采用衍生化- 串联质谱法检测尿液中氯胺酮及其代谢物去甲氯胺酮、脱氢去甲氯胺酮,获得了满意的结果。
本文通过对吸食氯胺酮麻醉药后的尿液,经衍生化-
gc-双质谱法检测氯胺酮原体及其代谢物的方法,由无锡市公安局刑警支队的沈笑平,何金海、窦莉等研究人员共同完成,现全文介绍如下:
实 验
1 仪器与试剂
美国瓦里安公司产CP -3800-Satam 2000型
气相一质谱仪(可以进行串联质谱分析),4000 r·minˉ1离心机(80- 2 B型,上海安亭科学仪器厂制造)。超声振荡提取器(JL-180型,上海杰理科技有限公司制造)。
氯胺酮对照品(中国药品生物制品检定所,按l00%汁算);醋酸甲酯、丙酸酐、无水碳酸钠、无水硫酸钠均为分析纯。
2 对照品溶液
氯胺酮对照品用醋酸甲酯配制成浓度为40 mg·mLˉ1的溶液。 已知该吸毒人员吸食氯胺酮的时间的尿液,按检材处理法处理后制成醋酸甲酯溶液。
3 检材的处理
取尿液2~3 mL于试管中,加人固体碳酸钠少量,加人丙酸酐约80μL(滴管约3~4滴),超声5min ,加人醋酸甲酯2 mL,超声萃取l0 min(超声器中放 200 mL烧杯.内放水,将试管放在烧杯中超声),离心(4000 r·minˉ1)3 min,取上层溶剂转入加有无水硫酸钠的4 mL样品瓶中,供
gc-MS/MS测定。
4
gc -MS/MS条件
4.1 色谱条件
进样室温度:280℃;柱程序升温:80℃(l min)以20℃/min升至280℃(4min),全程15 min;载气He流速:1 mL·minˉ1;分流比为:1:5,延时0.5 min后变为1:50,延时0.7min 后变为1:20。
4.2 质谱条件
连接杆温度:240℃;离子阱温度:150℃;阱盒温座:40℃。二级质谱灯丝能量:70uamps;一级质说谱灯丝能量:30 uamps 。电离方式:化学源(甲烷);最佳电离电流:0.6 volt;其余为仪器默认值。
5 MS/MS条件的优化
CI质谱中,分子离子峰信号明确,根据氯胺酮相对分子质量为237,CI质谱中往往存在M+l、M+2效应,所以准分子离子峰m/z为238、239。
去甲氯胺酮丙酰化物的分子离子峰m/z为280,脱氢去甲氯胺酮丙酰化物的分子离子峰rn/z为278。
在进行 MS/MS时,由AMD优化电离条件,确定最佳值以m/z238、m/z 280、m/z 278为母离子,获得特征性的分子离子碎片峰图,自建谱库。
6
gc-MS/MS质谱图
由于 CI是以正CI方式电离,对含有碳正离子结构的化合物响应信号强,灵敏度高,所以代谢物的衍生化时选用丙酸酐进行丙酰化,使代谢物结构特征明显。
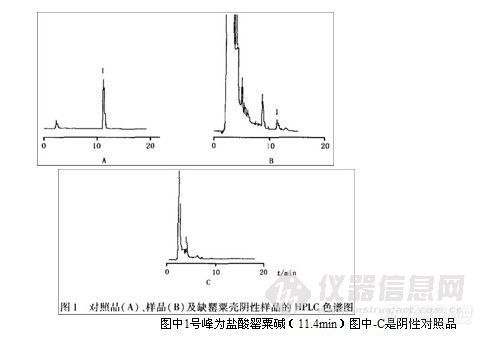
氯胺酮m/z 238(M+l)为母离子m/z 220为结构稳定的偶电子离子,故有较高的丰度,rn/z 207,179,152为一组,见图I -A。
去甲氯胺酮丙酰化物m/z 280或rn/z 281(M+1),m/z 224,207,179为一组,见图1 -B。
脱氢去甲氯胺酮丙酰化物m/z 278或m/z 279(M+1),m/z 222,205,177为一组,见图1—C。
![]()
7 检材提取的回收率
分别收集5名正常人尿液各l份,每份添加氯胺酮对照品40μg,在弱碱性条件下,进行测定,结果氯胺酮含量的平均回收率(n=5)为92.0%。由于是快速测定尿液中的氯胺酮及其代谢物,对于结合态的检测目标物来讲,提取率要低很多,但不影响在实案中的应用。
8 实际应用
案例l:对一男性吸毒嫌疑人的尿液,应用上述方法检测其中的毒品成分,1 h内就检出其尿液中含有氯胺酮、去甲氯胺酮(图2)。
![]()
![]()
9 结果与讨论
9.1 按本文方法,CC - MS/MS(CI离子源)最低检出限为0.05 ng,方法的最低检出浓度为:5×10-8 g·Lˉ1。本方法不仅可检验尿中原药氯胺酮,同时可检测尿中去甲氯胺酮、脱氢去甲氯胺酮,完全满足确证48 h以内是否吸食过氯胺酮的需要,其全部检验过程每份尿I h完成,检材尿仅需2~3 mL,达到了快速、灵敏、可靠的目的。
9.2 由于应用的是正离子Cl源,所以分子结构中电负性大的基团存在,不利于提高仪器的灵敏度,引入碳正离子,往往能提高灵敏度。另外考虑衍生化增加目标物的结构特征,提高定性的可靠性,所以选择丙酸酐形成丙酰化物。
9.3 由于胺类的化学特性,酰化过程在水溶液中都能瞬间完成,所以本方法采用在弱碱性条件下,直接将丙酸酐加人尿中,大大简化了检材的处理过程,既可快速测定,又不影响衍生化产物,但要注意的是,在弱碱性条件下的衍生化率高、如不显碱性则会影响衍生化效果。
9.4 由于串联质谱图要实验室自建谱库、所以必须对去甲氯胺酮、脱氢去甲氯胺酮进行确证。将氯胺酮水解,无法获得代谢物,所以只能根据质谱图的解析及碎片特征,用El与Cl方法确证已知吸食氯胺酮的吸毒者尿液中代谢物作为基准物,建立MS/MS谱图,同时也对正常人的尿液以及吸食海洛闪的人员的尿进行测定,排除干扰影响。
9.5 根据在临时检验中的应用可知,l0 h以内的尿液中都能检测到氯胺酮原药,10 h以后的尿液中就检测不出原药氯胺酮,只能检测出代谢物:去甲氯胺酮、脱氢去甲氯胺酮。随着时间的长短,两者之间的比例有所不同,两者量与时间的变化规律关系留待以后研究。
9.6 本方法将串联质谱改为一级质谱(Cl化学源)适用于尿中甲基苯丙胺、MDMA
![]()