获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
有关物质检查方法验证概要
项目 | 验证结果 |
流动相选择 | 0.05mol/L磷酸盐溶液(用0.05mol/L磷酸二氢钾溶液调节0.05mol/L磷酸氢二钠溶液pH值至7.0)-甲醇(40:60)。 |
专属性 | 各破坏条件下,产生的杂质与主成分能够有效分离,主成分峰未检出不纯物。 |
线性和范围 | 无相关研究内容。 |
定量限、检测限 | 雷贝拉唑钠的保留时间大约为9.3分钟,在±1分钟的时间范围计算基线噪音约为0.027,当S/N≒3时,雷贝拉唑钠的检测浓度为0.0125μg/ml,检测限为0.125ng,当S/N≒10时,定量限浓度为0.050μg/ml,定量限为0.50ng, |
准确度 | 无相关研究内容。 |
精密度 | 主峰面积和保留时间精密度RSD值(n=6)均小于2.0%。 |
溶液稳定性 | 室温放置8小时稳定性较好,主峰面积的RSD值为0.16%,归一化含量RSD值为0.13%,均小于2.0%。 |
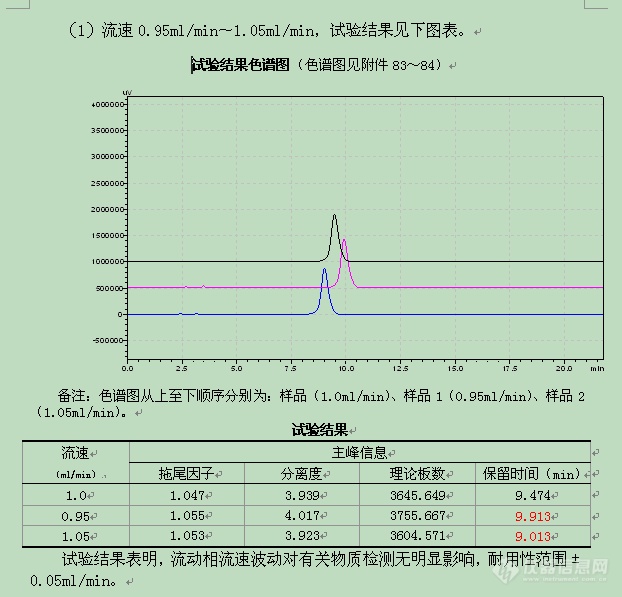
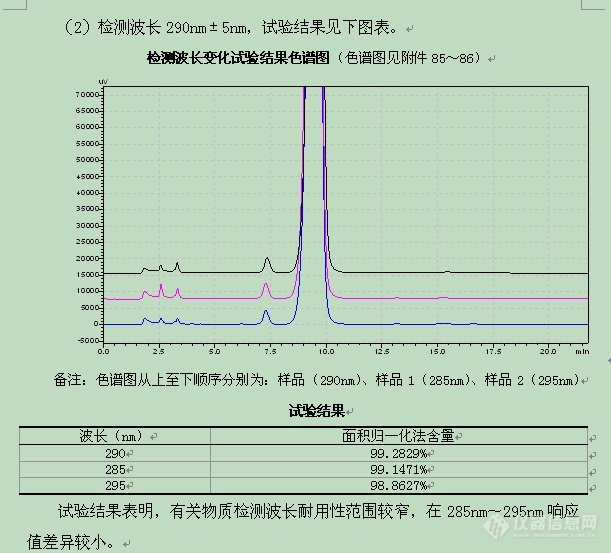
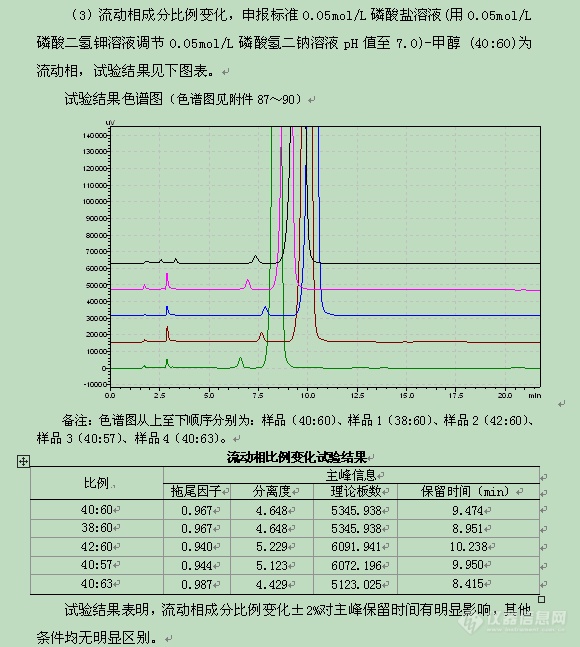
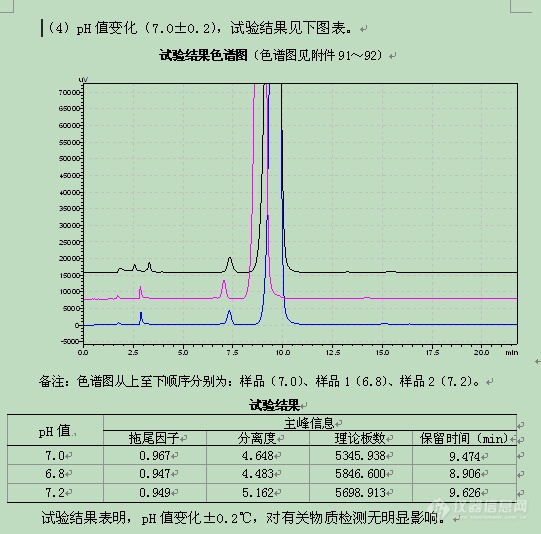
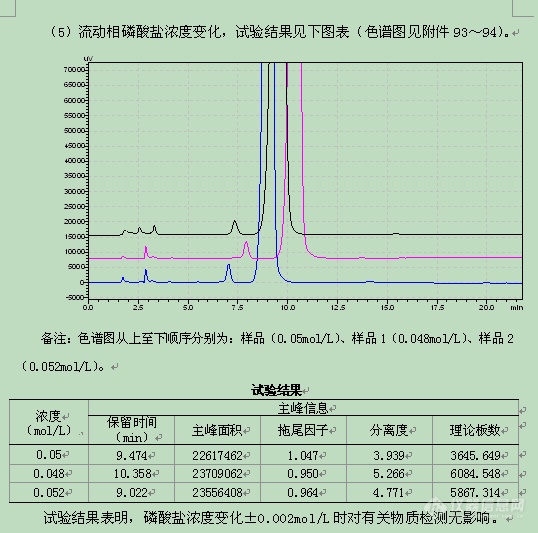
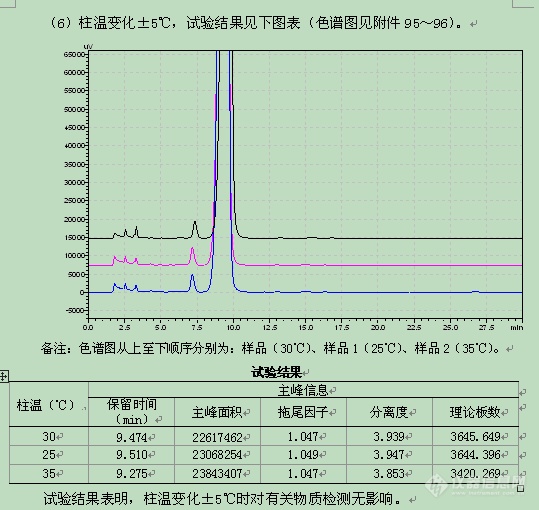
耐用性 | 流动相成分比例±2%;检测波长285nm~290nm;柱温为室温25℃~35℃,流速为1.0±0.05ml/min,pH值为7.0±0.2,磷酸盐浓度0.05±0.002mol/L。 |
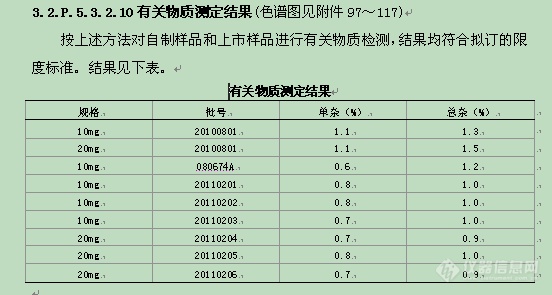
有关物质检查 | 上市品和自制品均符合规定。 |
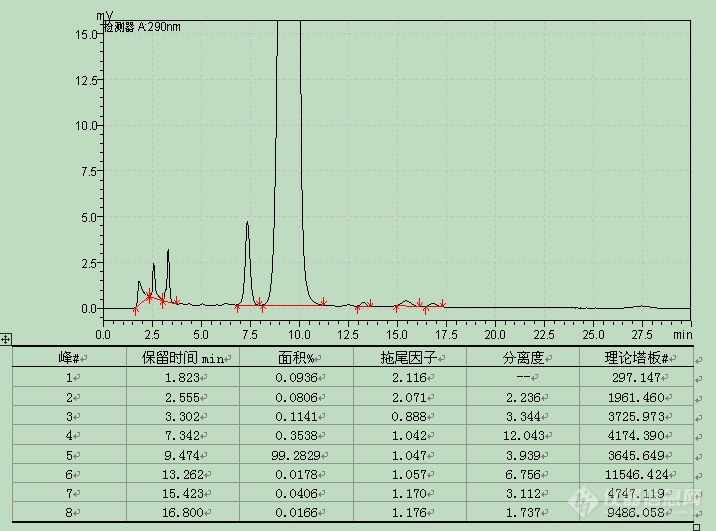
未破坏样品杂质情况
峰# | 1 | 2 | 3 | 4 | 5 |
保留时间(min) | 2.66 | 3.059 | 5.208 | 7.482 | 9.439 |
相对保留时间 | 0.28 | 0.32 | 0.55 | 0.79 | 1.00 |
210nm(%) | 0.126 | 0.1081 | 0.0962 | 1.1273 | 98.5423 |
290nm(%) | - | 0.0918 | - | 1.2049 | 98.7032 |
254nm(%) | - | - | - | 0.7608 | 99.2392 |
分离度 | - | 1.757 | 9.291 | 7.494 | 5.244 |
拖尾因子 | 0.845 | 1.353 | 1.077 | 1.076 | 1.053 |
理论塔板# | 1837.636 | 3520.617 | 6606.3 | 7290.177 | 9105.496 |
最大λ(nm) | 266 | 246/276 | 205/245 | 287 | 209/283 |
强光破坏样品杂质情况
峰# | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
保留时间(min) | 3.054 | 3.257 | 3.953 | 5.709 | 6.192 | 7.199 | 9.134 |
相对保留时间 | 0.33 | 0.36 | 0.43 | 0.63 | 0.68 | 0.79 | 1.00 |
210nm(%) | 0.2759 | 0.5703 | 0.128 | 0.0786 | 0.1375 | 1.218 | 97.5916 |
290nm(%) | 0.16 | 0.2205 | 0.1653 | 0.0837 | - | 1.3299 | 98.0406 |
254nm(%) | 0.2662 | 0.6454 | - | - | 0.2056 | 0.8122 | 98.0706 |
分离度 | - | 0.886 | 3.042 | 6.893 | 1.771 | 3.214 | 5.453 |
拖尾因子 | - | - | 0.854 | 1.172 | 1.195 | 1.112 | 1.09 |
理论塔板# | 2379.476 | 3987.047 | 3947.843 | 7859.452 | 7384.646 | 7232.524 | 9702.201 |
最大λ(nm) | 248/283 | 249 | 301/292/244 | 283/264/377 | 206/249 | 287 | 207/282 |
高温破坏样品杂质情况
峰# | 1 | 2 | 3 | 4 | 5 | 6 |
保留时间(min) | 2.619 | 2.788 | 3.13 | 6.362 | 7.568 | 9.584 |
相对保留时间 | 0.27 | 0.29 | 0.33 | 0.66 | 0.79 | 1.00 |
210nm(%) | 3.1381 | 1.3563 | 0.1235 | 0.0971 | 1.0838 | 94.2012 |
290nm(%) | 18.4139 | - | 0.438 | 0.0564 | 0.9933 | 80.0984 |
254nm(%) | 12.1392 | - | 2.5436 | - | 0.6564 | 84.6608 |
分离度 | - | 0.669 | 1.706 | 14.076 | 3.689 | 5.416 |
拖尾因子 | - | - | 1.157 | 1.115 | 1.107 | 1.094 |
理论塔板# | 1617.531 | 2125.215 | 6166.97 | 7204.525 | 7306.504 | 9657.418 |
最大λ(nm) | 311 | 226/263/307 | 252 | 206/246 | 287 | 207/282 |
氧化破坏样品杂质情况
峰# | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
保留时间(min) | 2.936 | 3.138 | 3.419 | 3.549 | 3.999 | 4.241 | 5.584 | 6.107 | 6.282 | 7.478 | 9.441 |
相对保留时间 | 0.31 | 0.33 | 0.36 | 0.38 | 0.42 | 0.45 | 0.59 | 0.65 | 0.67 | 0.79 | 1.00 |
210nm(%) | - | 0.3391 | 1.8783 | 0.828 | 1.5946 | 0.3247 | 0.5135 | 0.5983 | 0.2934 | 14.9764 | 78.6537 |
290nm(%) | 0.1205 | - | 0.9193 | - | 0.4124 | 0.1231 | 0.0995 | 0.4279 | - | 16.0782 | 81.8191 |
254nm(%) | 0.2792 | 0.1859 | 2.7772 | - | 0.5882 | 1.0845 | 0.2568 | 0.1743 | - | 10.6342 | 84.0197 |
分离度 | - | - | 0.532 | 0.491 | 1.879 | 1.156 | 4.39 | 1.192 | 0.183 | 1.352 | 5.024 |
拖尾因子 | - | - | - | - | 0.945 | - | - | - | - | 1.081 | 1.06 |
理论塔板# | 238 | 3136 | 2458 | 6924 | 5588 | 3367 | 2459 | 313 | 7327 | 7655 | |
最大λ(nm) | - | 276/250 | 208/251 | 259 | 223/277 /338 | 246/330 | 260/282 | 278 | - | 287 | 207/283 |
备注:各峰保留时间以杂质峰较多的210nm、290nm波长为主统计;“-”为未检出有关物质或不能计算。
(6)强酸破坏试验(色谱图见附件50~58)
酸破坏样品杂质情况
峰# | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
保留时间(min) | 3.316 | 3.586 | 4.080 | 4.732 | 5.129 | 6.320 | 7.310 | 8.548 | 9.290 | 11.466 | 14.045 | 21.165 |
相对保留时间 | 0.36 | 0.39 | 0.44 | 0.51 | 0.55 | 0.68 | 0.79 | 0.92 | 1.00 | 1.23 | 1.51 | 2.28 |
210nm(%) | 1.2108 | 3.0614 | - | 2.2711 | 5.2035 | 4.4185 | 1.1391 | 0.5429 | 75.9839 | 0.3785 | 0.7272 | 5.0631 |
290nm(%) | 0.6674 | 0.7963 | 0.2285 | 1.6037 | 0.7654 | 1.0644 | 1.3101 | 0.1359 | 87.0915 | 0.101 | 0.7949 | 5.4409 |
254nm(%) | 1.4461 | 3.8796 | - | 1.2177 | 5.9988 | 5.6217 | 0.6933 | 0.6965 | 75.0483 | 0.5087 | 0.6245 | 4.2648 |
分离度 | - | 1.418 | - | 4.766 | 1.426 | 4.163 | 3.133 | 3.375 | 1.809 | 4.532 | 4.709 | 10.499 |
拖尾因子 | 1.282 | 1.309 | - | - | - | 1.18 | 1.104 | 1.032 | 1.089 | 1.043 | 1.022 | 1.023 |
理论塔板# | 4465 | 5293 | - | 4441 | 5732 | 7039 | 7810 | 7175 | 8024 | 6998 | 10679 | 10852 |
最大λ(nm) | 208/250 /317 | 208/251 /326 | - | 208/305 /254 | 207/251 /348 | 206/247 | 287 | 207/251 | 204/283 | 207/251 | 284/249 | 284/249 |
碱破坏样品杂质情况
峰# | 1 | 2 | 3 | 4 | 5 |
保留时间(min) | 3.132 | 4.932 | 6.353 | 7.59 | 9.611 |
相对保留时间 | 0.33 | 0.51 | 0.66 | 0.79 | 1.00 |
210nm(%) | - | 0.1016 | 0.4201 | 1.1708 | 98.3076 |
290nm(%) | 0.4908 | 0.1639 | 0.1009 | 1.2573 | 97.9871 |
254nm(%) | - | - | 0.5578 | 0.7986 | 98.6437 |
分离度 | - | - | 4.935 | 3.623 | 5.56 |
拖尾因子 | - | 0.791 | 1.075 | 1.095 | 1.092 |
理论塔板# | - | 6704.387 | 5722.093 | 7724.549 | 10196.93 |
最大λ(nm) | - | 281 | 206/247 | 287 | 206/282 |
专属性杂质谱统计
试验项目 | 相对保留时间(RRT) | ||||||||||||||||
0.28 | 0.29 | 0.33 | 0.36 | 0.39 | 0.42 | 0.44 | 0.51 | 0.55 | 0.63 | 0.68 | 0.79 | 0.92 | 1.00 | 1.23 | 1.51 | 2.28 | |
未破坏样品 | √ | - | √ | - | - | - | - | √ | - | √ | - | √ | - | - | - | ||
强光破坏样品 | - | - | √ | √ | - | - | √ | - | - | √ | √ | √ | - | √ | - | - | - |
高温破坏样品 | √ | √ | √ | - | - | - | - | - | - | - | √ | √ | - | √ | - | - | - |
氧化破坏样品 | - | √ | √ | √ | √ | √ | √ | - | √ | √ | √ | √ | - | √ | - | - | - |
酸破坏样品 | - | - | - | √ | √ | - | √ | √ | √ | - | √ | √ | √ | √ | √ | √ | √ |
碱破坏样品 | - | - | √ | √ | √ | - | √ | √ | - | - | √ | √ | - | √ | - | - | - |
专属性试验物料平衡统计
试验项目 | 称样量(g) | 稀释倍数 | 总峰面积 | 主峰面积归一化法含量 | 总峰面积 | 平衡率 |
与浓度比值 | ||||||
未样品 | 0.6549 | 50 | 16515834 | 98.703% | 1260943197 | 100.00% |
强光破坏样品 | 0.6588 | 50 | 17553365 | 98.041% | 1332222602 | 105.65% |
高温破坏样品 | 0.6567 | 50 | 17352531 | 80.098% | 1321191640 | 104.78% |
氧化破坏样品 | 0.6303 | 50 | 14666784 | 81.819% | 1163476440 | 92.27% |
酸破坏样品 | 0.5801 | 50 | 13273905 | 87.091% | 1144104896 | 90.73% |
碱破坏样品 | 0.6541 | 50 | 16917104 | 97.987% | 1293158844 | 102.55% |
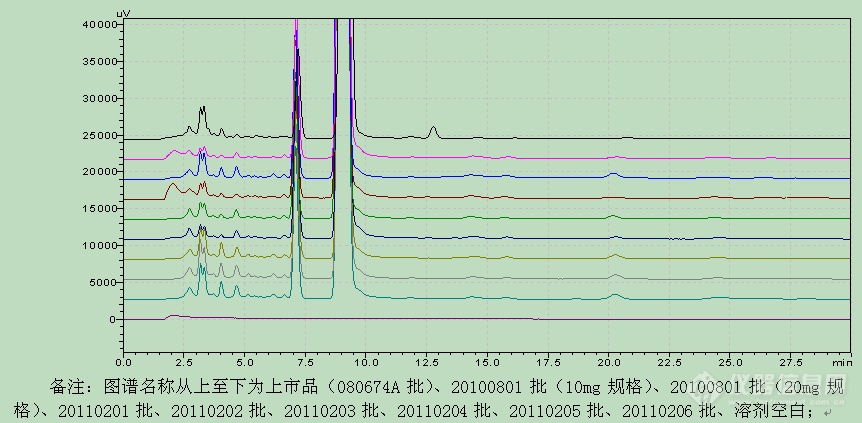
中试样品和上市品色谱图(UV 290nm)
批号 | 0.23 | 0.30 | 0.36 | 0.37 | 0.45 | 0.52 | 0.60 | 0.70 | 0.73 | 0.78 | 1.00 | 1.31 | 1.40 | 1.76 | 2.27 |
80674 | - | √ | √ | √ | √ | √ | √ | - | √ | √ | √ | √ | - | √ | |
20100801(10mg) | √ | √ | √ | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ |
20100801(20mg) | - | √ | √ | √ | √ | √ | - | √ | √ | √ | √ | - | - | √ | √ |
20110201 | - | - | √ | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ |
20110202 | - | - | √ | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ |
20110203 | - | - | √ | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ |
20110204 | - | - | √ | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ |
20110205 | - | - | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ | |
20110206 | - | - | √ | √ | √ | √ | - | - | √ | √ | √ | - | - | √ | √ |
检测限的确定
序号 | 峰高(mV)εmax | 峰高(mV) εmin | 峰高(mV) Δε | 平均峰高(mV) Δε | 检测限(mV) |
1 | 0.002 | 0.007 | 0.009 | 0.009 | 0.027 |
2 | 0.004 | 0.005 | 0.009 |
检测限验证浓度
序号 | 稀释倍数 | 浓度(µg/ml) | 进样量(ng) | 峰高(mV) | 平均峰高(mV) | S/N |
1 | 100 | 10.00 | 100.00 | 14.510 | - | |
2 | 10000 | 0.100 | 1.00 | 0.158 | - | |
3 | 20000 | 0.050 | 0.50 | 0.104 | - | 11.6 |
4 | 80000 | 0.0125 | 0.125 | 0.026 | 0.029 | 3.2 |
4-1 | 80000 | 0.0125 | 0.125 | 0.033 | ||
4-2 | 80000 | 0.0125 | 0.125 | 0.028 |
1%自身对照溶液精密度试验结果
项目 | 1 | 2 | 3 | 4 | 5 | 6 | 平均值 | RSD |
峰面积 | 210203 | 210451 | 210227 | 210652 | 210851 | 210749 | 210522 | 0.13% |
保留时间 | 8.911 | 8.955 | 8.964 | 8.957 | 8.953 | 8.945 | 8.948 | 0.21% |
溶液稳定性试验结果
时间(h) | 0 | 2 | 4 | 6 | 8 | 平均值 | RSD |
主峰面积 | 22652515 | 22663551 | 22676001 | 22706635 | 22745063 | 22688753 | 0.16% |
归一化含量(%) | 98.148 | 98.074 | 97.995 | 97.910 | 97.830 | 97.991 | 0.13% |
杂质个数 | 14 | 11 | 11 | 11 | 11 | - | - |
耐用性研究项目 | 试验方法的条件 | 确认的耐用性范围 |
流动相成分比例 | 0.05mol/L磷酸盐溶液(用0.05mol/L磷酸二氢钾溶液调节0.05mol/L磷酸氢二钠溶液pH值至7.0)-甲醇 (40:60) | ±2% |
流速(活性成分的保留时间变化) | 1.0ml/min | ±0.05ml/min |
色谱柱温度 | 30℃ | ±5℃ |
检测波长 | 290nm | 285nm~290nm |
流动相缓冲液pH值 | 7.0 | ±0.2 |
流动相缓冲液磷酸盐浓度 | 0.05mol/L | ±0.002mol/L |