问题:峰形后拖尾是什么原因造成的?
答案:色谱柱有物理损坏是造成峰形拖尾的根源。唯一的解决方法就是更换新柱。
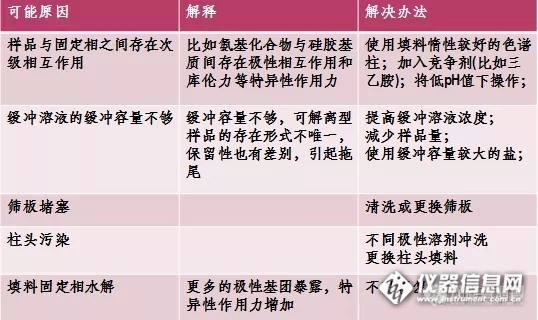
流动相和样品中的杂质是色谱柱主要污染源。流动相所用的各种溶剂至少是分析纯,尽量使用色谱纯试剂。流动相所用的水最好是超纯水或全玻璃器皿双蒸水。用前先用0.45um溶剂微孔过滤(器)过滤,除去可能存在的微粒。流动相建议现用现配,对于含盐的溶液尤其注意长置会产生细菌或出现沉淀。此外还得保证储存流动相的容器清洁。
复杂样品可选用0.45um溶剂微孔过滤(器)或样品预处理柱对样品进行预处理,确保样品中不含微粒杂质。若样品不便处理,要使用保护柱。柱内填料污染时,可将柱头螺丝卸下,用专用工具挖出柱内前段被污染填料,再以相同的填料重新填入,修复。或以能溶解污染物的流动相按色谱柱使用的相反方向,冲洗色谱柱(约20至30倍柱体积,或视具体情况而定;另外,此时不能接检测器),将污染物冲出色谱柱,再按色谱柱标明的使用方向使用。
当断定是柱进口处有异物时,可将柱头螺丝卸下,取出滤帽并将其置于20%硝酸溶液中,用超声波清洗约20分钟,再置于超纯水中,并用超声波清洗约10分钟,重新装入色谱柱内即可。
样品在柱上超载能引起峰展宽,拖尾(或伸舌)。适当减小进样量或样品浓度(需要时,可提高检测器灵敏度)直至峰形和保留时间不再改变,便可消除这种不良影响。减小进样量还能改善其分离度。正常情况下,样品中每种化合物在150×4.6mm柱中进样量保持在3~50ug范围内,不会引起明显超载。
选择合适的样品溶剂,以排除不必要的干扰。最好选用流动相溶解样品。
即进样阀、色谱柱及检测器间的管路过长、直径过粗、管路接头不匹配、有死体积,是影响色谱柱柱效的主要因素之一。所以在可能的条件下,色谱柱的两端连接管路要尽可能短,连接管内径尽可能细小,切口务必平整光滑,尽可能的减小死体积,以防止因样品扩散造成不能反映色谱柱真实柱效等情况发生。
在缓冲不好或离子强度过低的分离中,也会出现保留时间重现性差和峰形拖尾。增加缓冲浓度(与样品大小相匹配)能改善此情况。
仔细分析色谱柱内填料表面性质可知:反相色谱柱填料的表面大约被键合相覆盖了一半,其余的就是未被键合的硅醇基团。所谓的保留是指样品分子与键合相相互作用的结果。但是,酸性或碱性化合物与硅胶表面残存的硅醇基团或金属杂质之间相互作用而引起双重保留机理,因此,发生了峰形拖尾。为了减小峰形拖尾,通常可在流动相加入25mM三乙胺(抑制剂)。三乙胺能与硅醇基团发生强烈的相互作用,从而降低或阻断样品分子与硅醇基团的作用,以保证保留机理正常进行,并大大缓解了峰形拖尾。使用长链的硅醇基抑制剂,虽起效较慢,但持续力较三乙胺长。因为抑制剂分子中除了胺基与硅醇基团发生极性作用外,大分子中非极性部分与固定相也会发生反相作用而产生保留现象。如果将抑制剂加至样品中,效果会显著。
柱内金属污染(Fe,Ni等)可引起某引些化合物峰形拖尾。金属可由填料本身带进,也可由腐蚀性流动相缓慢溶解柱进口的金属滤片,随流动相沉积到柱填料中。例如C18柱填料被金属污染后,造成酸性/碱性样品拖尾,可用碱洗/酸洗后再键合的填料,得到的峰是尖锐且对称的。 =======================================================================
【
活动内容】
1、每个工作日上午10:00左右发布一个色谱问答题,版友根据题目给出自己理解的答案。
2、每个工作日下午15:10公布参考答案。
【
活动奖励】
幸运奖:抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励
2钻石币(抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);
中奖名单:
yifan1117(注册ID:yifan1117)
大川之子,纵横四海(注册ID:chuangu120)
999youran(注册ID:999youran)
zengzhengce163(注册ID:zengzhengce163)
dahua1981(注册ID:dahua1981)
![]()
![]()
【
注意事项】同样的答案,每人只能发一次
PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。
下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。