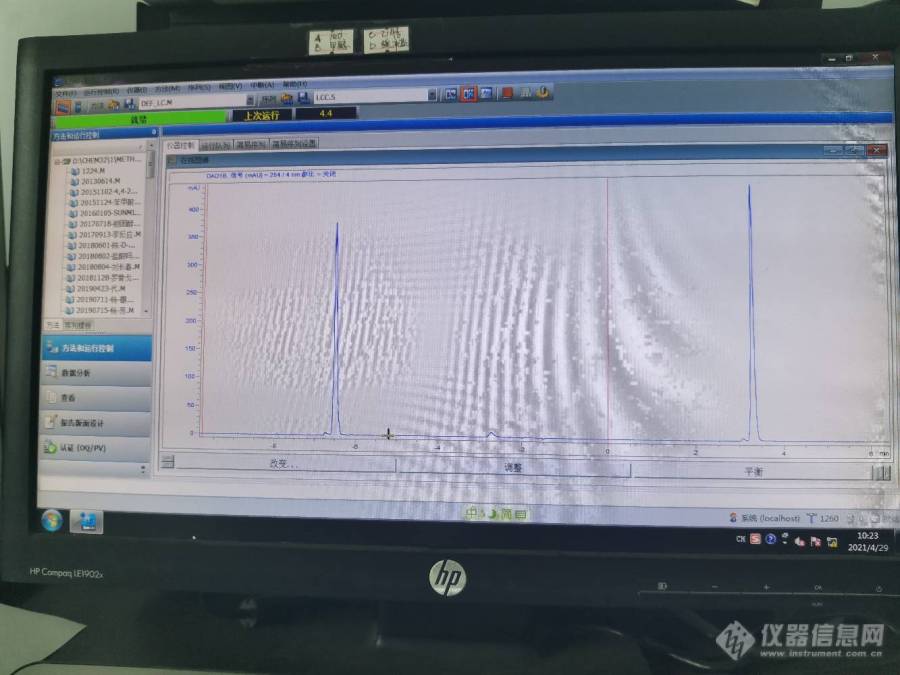
高效
液相色谱分析中进样基线很好,保留时间能锁定,压力也稳定,但是峰面积差别很大,大概有2倍的差别,这种情况出现的原因可能是多方面的,建议采用以下方法解决:1调换一根新色谱柱,如果情况一样,则排除柱子的原因。(推荐使用赛分色谱柱)
2将在线过滤器拆下,用超声波清洗,如果结果一样,说明与在线过滤器无关。
3考虑是不是进样阀有问题或者定量环堵。建议多进几针样品,看是否有规律,如果没有规律,应检查一下进样器的其他部位,如果是进样器系统出现问题,应尽快解决之。
4对进样器清洗一下,清洗干净后先进一针空白,再进样,看看结果如何,如果有明显减少,那可能是进样器污染,有样品残留在进样阀体内,在切换的过程中被流动相带入色谱柱。
5考虑样品溶液是否均匀。建议同一个浓度连续进样5次,看一下结果如何变化,有没有规律;如果是样品溶液不均匀,可以再搅拌一下。
6考虑光源是不是正常。
7考虑浓度是否在线性范围内。有时候定量时浓度太高或太低都会产生较大的分析误差。
8考虑取样方式是否正确,每次取样后是否对针头外面的附着液进行清除。
9高效
液相色谱仪的计算机软件编制可能存在问题。对比可以适当改变软件。