维权声明:本文为Ins_a80bbdc7原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
免脱盐柱除盐的蛋白组学前处理方法
季学猛,史爱莹,张 燕,王 硕
(南开大学 医学院, 天津 300071)
摘 要:现有的蛋白组样品前处理中脱盐柱除盐的方法存在步骤繁多、易损失微量样品等缺点。这里,一种超滤管辅助酶解及加热潮解除盐的蛋白组学前处理方法被提出。具体而言,是利用超滤管辅助,通过离心在酶解前将缓冲液置换成碳酸氢铵溶液,酶解后多肽溶液中的碳酸氢铵被加热除去,最终得到脱盐的纯净多肽。该方法操作简便,无需洗脱步骤,可以方便地与
LC-MS直接衔接,而且可以减少前处理引起的氨基酸残基去酰胺和氧化修饰,有利于蛋白组学及其蛋白修饰分析。
关键词:蛋白组学;样品前处理;脱盐柱; 加热; 碳酸氢铵
中图分类号:O657.63 文献标识码:
An Improved Proteomic Pretreatment Method without Desalination Column
JI Xuemeng,SHI Aiying,ZHANG Yan,WANG Shuo
(School of Medicine, Nankai University, Tianjin 300071, China)
Abstract: The existing method for desalting protein samples in pre-processing is associated with several drawbacks, including a high number of procedural steps and the potential loss of minute sample quantities. In this context, a novel protein sample pre-processing method is proposed, which utilizes ultrafiltration tubes to assist in enzymatic digestion and employs heat-induced desalting. Specifically, the ultrafiltration tubes are employed to facilitate the replacement of the buffer solution with an ammonium bicarbonate solution via centrifugation before enzymatic digestion. Subsequently, following enzymatic digestion, the ammonium bicarbonate within the peptide solution is removed through a heating process, ultimately yielding desalted and pure peptides. This method offers simplicity in operation, eliminates the need for elution steps, enables seamless integration with
LC-MS, and serves to mitigate the formation of amino acid residue deamidation and oxidation modifications that may be induced during pre-processing, thereby benefiting both proteomics and protein modification analyses.
Key words: Proteomic analysis; Sample pre-treatment; Desalting column; Heating; Ammonium bicarbonate
通过高效
液相串联质谱对生物系统中全蛋白进行定性定量,已经成为一种主流的分析工具。通过高效
液相串联质谱对生物系统中全蛋白进行定性定量,已经成为一种主流的分析工具。大规模的蛋白组研究依赖于蛋白被切割成肽段,然后进行后续的定性定量分析。目前,蛋白组学样品前处理方法通常使用胰蛋白酶将蛋白质样品切割成多肽。胰蛋白酶的最适作用pH为7.5-8.5
[1],碳酸氢铵是质谱预处理中最常用到的一种盐,50mM碳酸氢铵水溶液可以提供酶解过程需要的弱碱性环境。然而,碳酸氢铵在酶解过程中大量存在,容易影响后续质谱分析。目前常用反相C18树脂去除盐和缓冲液
[2-4]。多肽在高水相流动相中与反相柱结合,盐和缓冲液被洗去,然后用高有机相流动相洗脱多肽。然而,多肽对C18吸附性有差异,包括磷酸化多肽在内的亲水肽可能不能与C18树脂很好地结合,疏水性强的多肽与C18树脂可能结合牢固,不易被洗脱
[5-7],这些问题可能导致样品的损失;而且,用来洗脱多肽的有机溶剂还需进一步通过真空离心干燥去除,极性较小的有机溶剂还可能溶解塑料制品中聚合物,造成多肽样品的聚合物污染,导致质谱峰偏移
[8,9]。这些问题可能导致蛋白质检出种类减少、重复性差、甚至无检出信号等问题。基于这种临床和科研上的需求, 需要对蛋白组学样品前处理方法进行改进。
1? 实验原理
蛋白质组学是后基因组时代的产物。基因需要依赖于转录和翻译后后的产物蛋白质行使功能。基因组是固定不变的,而蛋白质组会响应环境的变化。因此,同一生物在生物体不同部位、生命的不同时期以及不同的环境中,具有不同的蛋白质表达。人类基因组测序计划的完成并没有给人提供解开生命的密钥,科学家把兴趣转到蛋白质,希望通过蛋白质组的研究来进一步解开生命的本质。
目前鸟枪法是蛋白质组学分析应用最广的分析策略
[10-12]。该方法先将蛋白酶解成肽段,然后通过色谱分离肽段混合物,再用质谱的电喷雾电离(ESI)技术将肽段碎裂,根据碎裂谱图的离子峰信息进行数据库搜索来鉴定肽段,最后将鉴定的肽段进行组装、重新归并为蛋白质
[13]。
电喷雾电离具体包括以下几个过程:样品首先通过一个毛细管喷针被喷出来,进入质谱仪,在喷针的外面用鞘气加热样品,辅助样品的雾化。加热雾化过程中溶液中的流动相或者溶剂挥发,剩下的气态离子在毛细管喷针尖端被电离。这些离子在质谱仪入口处被真空抽到质谱仪里,电场驱动进入质谱仪进行分子量的检测
[14]。
一般来说,电喷雾质谱法对盐类的容忍度较低。一方面是因为小分子盐类在电喷雾系统中存在较强的竞争性电离效应,从而导致强烈的离子抑制效应,使待测物的灵敏度明显降低
[15]。其次,盐类的存在会产生一系列的离子加成峰,这使得谱图的解析变得复杂
[16-18]。此外,过多的盐分会腐蚀和污染质谱系统的硬件,严重时导致硬件损坏,需要及时清洗
[19]。因此,蛋白组学样品前处理过程中,脱盐步骤是非常必要的。
2? 存在问题
为减少机器的损坏,并且提高检测灵敏度,上机前一般都会要求对蛋白组学样品脱盐处理。目前脱盐常用的方法是反相C18树脂脱盐柱法。脱盐柱法分为结合、清洗、洗脱三步。首先,利用疏水相互作用,多肽在高水相流动相中与反相柱结合。然后,使用水反复清洗盐和缓冲液。最后,用有机溶剂破坏多肽与C18的结合,洗脱多肽。然而,该方法存在一系列的弊端,比如多肽对C18吸附性有差异,包括磷酸化多肽在内的亲水性比较强的多肽可能不能与C18树脂很好地结合,会造成结合阶段的样品损失,而疏水性强的多肽与C18树脂结合牢固,洗脱阶段却难以洗脱下来,所以亲水性强的多肽和疏水性强的多肽都会在脱盐柱脱盐过程中损失,进而导致样品的选择性偏好,会损坏质谱结果的客观性。而且,最终多肽样品上机前需要在0.1%甲酸溶液中溶解,因此,有机溶剂洗脱后的多肽不能很好的与
LC-MS衔接,还需真空离心干燥去除有机溶剂。使用有机溶剂洗脱样品可能会造成样品的PEG聚合物污染,导致质谱峰偏移。这些问题导致脱盐柱脱盐后的蛋白组学样品重复性差、检出蛋白质种类低问题,因此亟需一种脱盐柱脱盐的替代方法。
3? 改进措施
将酶解前超滤管中的缓冲液通过反复离心置换成碳酸氢铵水溶液。酶解后利用碳酸氢铵加热潮解的特性除去多肽溶液中的碳酸氢铵成分,最终得到纯净的多肽。多肽样品溶解在0.1%甲酸溶液中后可以直接上机检测。
4? 改进后效果
首先取100μg蛋白样品,用6 M 盐酸胍稀释至400μL。加入10μL 500mM 三-(2-羧乙基)膦、 8 μL 1M 碘乙酰胺,混匀。室温避光振荡反应 40 min。将样品转移至10KD超滤管,室温10000g离心30分钟。加入400 μL 50mM碳酸氢铵溶液置换5次。用200 μL 50 mM 碳酸氢铵溶液垂悬,加入2μg胰蛋白酶,于37 ℃酶解过夜。10000g离心10分钟,收集滤液。真空离心干燥滤液,干燥后观察管底,如有白色盐粉末,加入少量超纯水溶解白色盐,转入75℃金属浴烘干,碳酸氢铵在烘干过程中分解挥发。上机前加入50μL 0.1% 甲酸溶液,装入超洁净样品瓶,上机检测。
4.1 潮解脱盐效果评估
利用高效
液相色谱(HPLC)分析表明,在五次洗涤步骤后 ,三-(2-羧乙基)膦和碘乙酰胺的浓度至少降低了100000倍,并且降至检测阈值以下,表明五次连续洗涤足以将杂质降低到不干扰的水平(
图1A)。酶解后,离心
收集到滤液200μL。试纸法测滤液pH,呈弱碱性(pH 7.5-8.0)。多肽样品经过真空离心干燥后,观察管底,可见白色盐粉末,如
图1B所示。加入2μL 超纯水溶解白色盐,转入75℃金属浴烘干,碳酸氢铵在烘干过程中分解挥发,白色粉末消失,如
图1C所示。用200
μL超纯水复溶多肽,测pH为7.0。这些结果说明碳酸氢铵被去除。
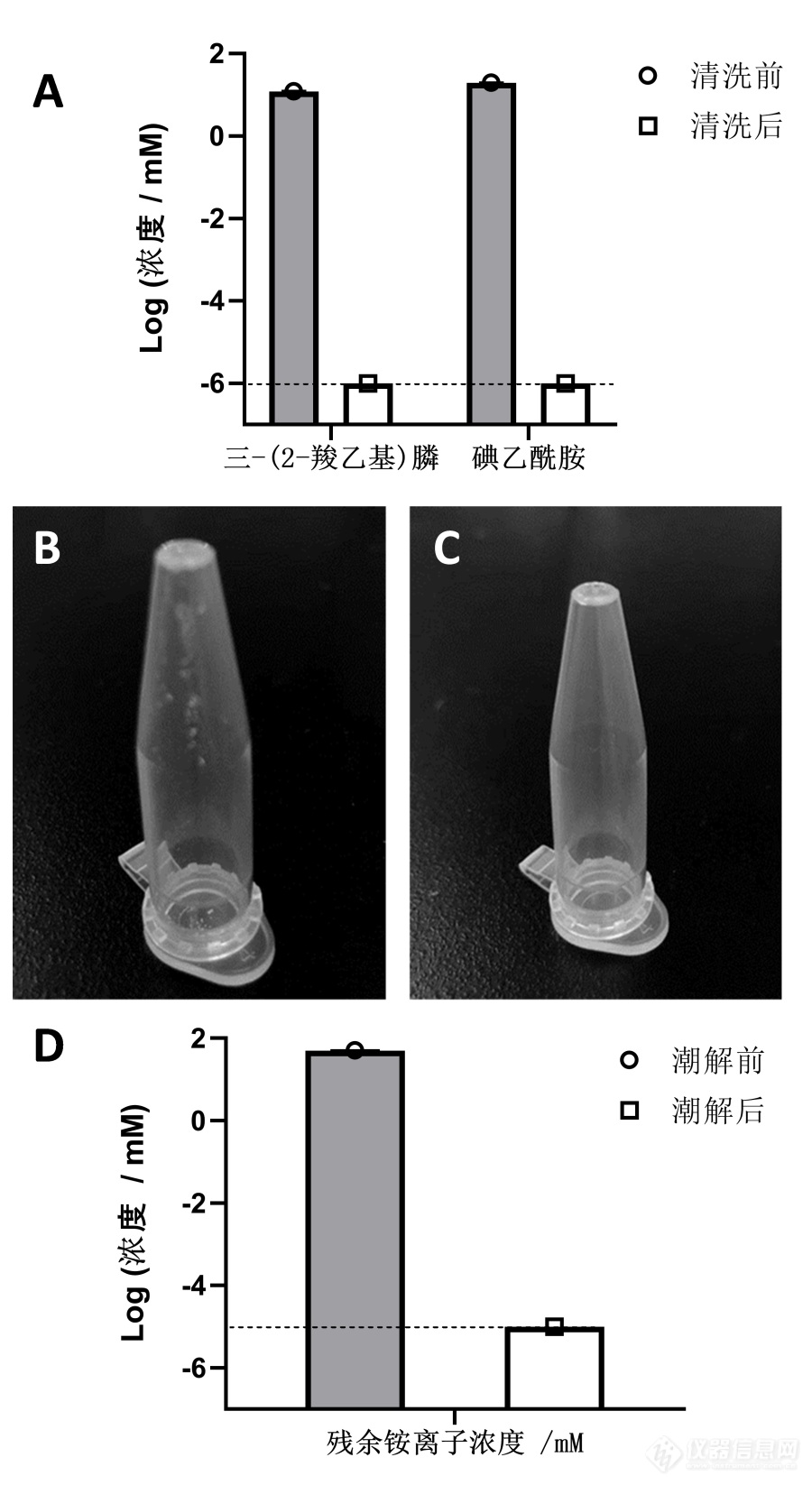
![]() 图1 脱盐的效果与验证。A:高效液相色谱(HPLC)分析图;B:多肽干燥后可观察到管壁上残留白色盐分;C:加热碳酸氢铵潮解后管壁上白色盐分消失; D:潮解前后的氨离子浓度对比图。数据来自三次独立重复实验(n = 3)。
图1 脱盐的效果与验证。A:高效液相色谱(HPLC)分析图;B:多肽干燥后可观察到管壁上残留白色盐分;C:加热碳酸氢铵潮解后管壁上白色盐分消失; D:潮解前后的氨离子浓度对比图。数据来自三次独立重复实验(n = 3)。为了直接确认碳酸氢铵的去除,采用分光光度尼斯勒试剂法对氨浓度进行定量。分光光度尼斯勒试剂法包括以下步骤:将样品用去离子水稀释,向样品中加入一定量的分光光度尼斯勒试剂,封闭瓶子并摇动,让混合物静置5-10分钟,使用分光光度计在紫外-可见光谱范围内的波长630nm处测量吸光度,通过相同方法处理一系列氨标准溶液,测量吸光度值并绘制标准曲线,最后,通过使用样品的吸光度值查找标准曲线上相应的氨离子浓度来计算样品浓度。实验结果显示氨离子浓度从50 mM降低到不到10 nM,证实了通过热力潮解去除碳酸氢铵的有效性(
图1D)
4.2 新方法对不同分子量的蛋白质进行的蛋白质回收性能的研究
蛋白质的分子量分布范围很大,从几千到几十万道尔顿不等。为了研究在酶解之前,超滤管辅助酶解及加热潮解除盐的蛋白组学前处理方法(以下称为新方法)在蛋白质回收方面是否存在偏好,在酶解前比较了溶液法(in-solution)、过滤辅助样品制备法(Filter-Aided Sample Preparation, FASP) 和本研究提出的新方法中的蛋白质回收率。蛋白质定量采用了与还原剂相容的BCA蛋白质测定试剂盒,吸光度在分光光度计(Agilent Technologies,美国)上以562纳米的波长读取。在本研究中测试的蛋白质包括细胞色素C(约12.4 kDa)、绿色荧光蛋白(GFP)(约27 kDa)、抗凝血酶III(约58 kDa)、转铁蛋白(约80 kDa)、免疫球蛋白G(IgG)(约150 kDa)和纤维连接蛋白(具有广泛的分子量范围,通常超过200 kDa)。实验进行了三次重复,并使用小提琴图来呈现结果。
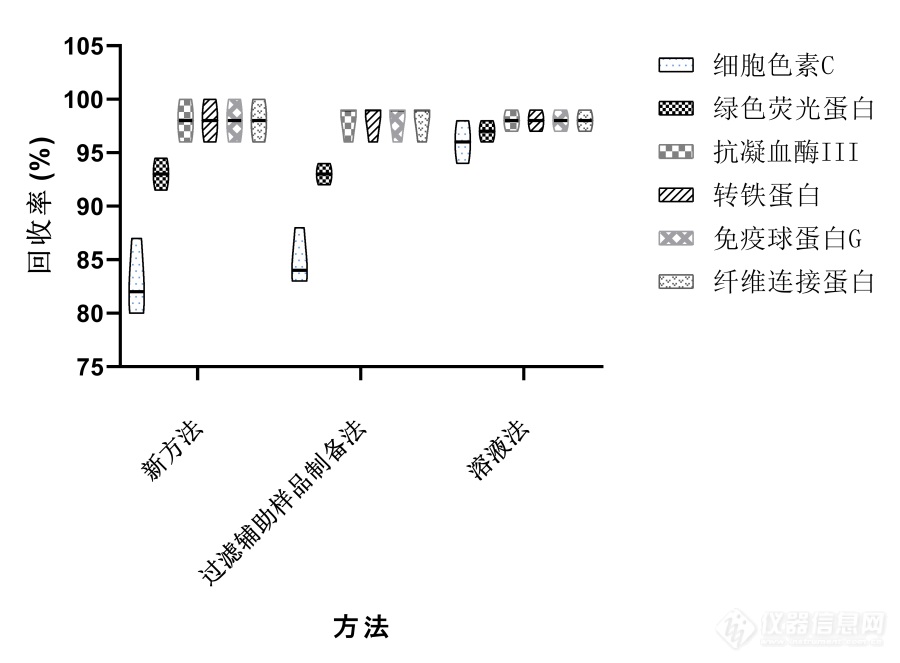
![]() 图2使用溶液法、过滤辅助样品制备法和新方法对不同分子量的蛋白质进行的蛋白质回收率评估图。
图2使用溶液法、过滤辅助样品制备法和新方法对不同分子量的蛋白质进行的蛋白质回收率评估图。结果清晰地表明,在不同分子量的蛋白质中,无论是在溶液法、过滤辅助样品制备法还是新方法中,所有三种方法在蛋白质回收率方面均达到了80%以上(图2)。总的来说,在所测试的分子量范围内,过滤辅助样品制备法和新方法之间的蛋白质回收率没有显著差异。然而,值得注意的是,对于低分子量蛋白质,溶液方法展现了略高的回收率,这可能归因于使用了10 kDa分子量截止滤器。 具体来说,对于低分子量蛋白质细胞色素C,过滤辅助样品制备法和新方法的平均回收率分别为85%和83%,而溶液方法的回收率为96%。然而,随着蛋白质分子量的增加,过滤辅助样品制备法和新方法的回收率显著提高。例如,对于分子量约为27 kDa的绿色荧光蛋白(GFP),过滤辅助样品制备法和新方法的回收率约为93%,而溶液方法对GFP蛋白的回收率为97%。此外,对于分子量超过50 kDa的较大蛋白质,如抗凝血酶III,使用过滤辅助样品制备法和新方法的回收率与溶液方法没有显著差异,均达到约98%。可见,本发明方法在蛋白质酶解之前对不同分子量的蛋白质回收率能达到83%至98%范围,达到了较好的蛋白截流作用。
4.3新方法中潮解脱盐对不同大小和亲水性的肽段回收性能的研究
为了探究不同方法酶解后对不同分子量的肽段进行潮解除盐的潜在偏好,对潮解法脱盐和C18脱盐的多肽回收率进行比较分析。在脱盐过程之前和之后,使用高效
液相色谱(HPLC)测量了肽段的浓度。使用了一个反相柱,并监测了220 nm处的吸光度。受测试的肽段包括亮氨酸脑啡肽(分子量:555.68 Da,由五个氨基酸组成)、血管加压素(分子量:1084.23 Da,由九个氨基酸组成)、生长抑素(分子量:约1637.89 Da,由十四个氨基酸组成)和胰高血糖素(分子量:约3483.87 Da,由二十九个氨基酸残基组成),三次重复实验的结果呈现在小提琴图中(图3A)。结果显示,通过C18柱脱盐得到的回收率在75%到90%之间,与肽段的分子量之间没有明显的相关性。这可能是由于在C18柱脱盐过程中样品损失的因素,比如柱子的结合性能和洗脱效率。相比之下,潮解脱盐在所有测试的肽段中实现了超过96%的回收率,而不受它们的分子量影响。这一结果表明,潮解脱盐在回收不同分子量的肽段方面非常有效。
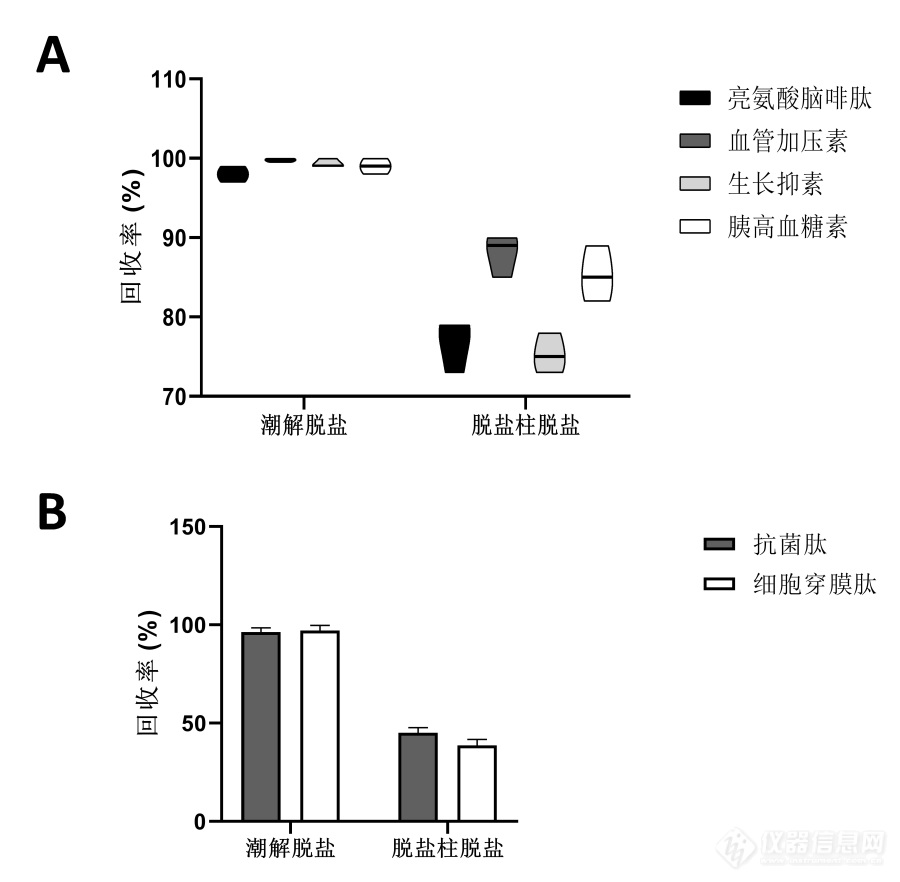
![]() 图3肽段回收性能比较。A:使用两种脱盐方法(潮解去盐和脱盐柱脱盐)评估不同分子量的肽段回收率;B:使用潮解去盐和脱盐柱脱盐对高亲水性和疏水性肽段进行比较回收分析;实验均进行了三次独立的重复。
图3肽段回收性能比较。A:使用两种脱盐方法(潮解去盐和脱盐柱脱盐)评估不同分子量的肽段回收率;B:使用潮解去盐和脱盐柱脱盐对高亲水性和疏水性肽段进行比较回收分析;实验均进行了三次独立的重复。接下来,使用C18脱盐柱脱盐和潮解脱盐对高亲水性和疏水性肽段的回收率进行了比较分析。选择了抗菌肽(Cat. No. LL37-05MG)和细胞穿膜肽(CAS号:697226-52-1)分别代表疏水性和亲水性肽段。结果显示,脱盐柱脱盐对抗菌肽和细胞穿膜肽的回收率相对较低,平均回收率分别为45%和39%。相比之下,新方法潮解除盐后对这两种肽段都实现了超过95%的回收率,而不受它们的亲水性影响(图3B)。这些发现表明,脱盐柱脱盐中的回收率受到肽段亲水性的影响。这可能归因于脱盐柱脱盐依赖于疏水相互作用将肽段结合到C18柱树脂上,然后使用亲水溶剂进行洗脱,这可能导致不同亲水性的肽段被选择性保留。另一方面,潮解脱盐提供了原位操作的优势,从而在脱盐过程中避免了肽段的损失。我们的研究结果表明,潮解脱盐不会导致肽段的损失,并实现了比C18柱脱盐更高的多肽回收率。
4.4 新方法在实际蛋白质组学分析中的应用比较
小鼠肠组织用PBS磷酸盐缓冲液润洗两次,以去除任何残留物质,然后与蛋白酶抑制剂混合。将组织使用组织研磨仪进行破壁,然后在冰浴中使用超声波(200W)裂解,直到悬浮液变清澈。通过孔径为0.22μm的微孔滤器纯化得到裂解物。使用BCA分析试剂盒测定蛋白质浓度。随后,采用三种方法进行了酶解:溶液法、过滤辅助样品制备法和新方法
(蛋白组学原始数据保存于PXD044209)。
酶解后,使用高性能
液相色谱将蛋白质分离,并使用QE质谱仪进行分析。具体来说,酶解后的肽样品采用Q Exactive Plus质谱仪联用EASY nano
液相色谱系统进行分析,该系统配备了EASY纳米电喷雾接口。色谱装置包括Pepmap纳米捕获柱(C18,5 μm,100 ?,100 μm × 2 cm)和EASY-Spray柱(Pepmap RSLC,C18,2 μm,100 ?,50 μm × 15 cm)。在色谱梯度中使用溶剂A(0.1%甲酸)和溶剂B(80% CH3CN/0.1%甲酸),梯度如下:0–8% B持续3分钟,8–28% B持续42分钟,28–38% B持续5分钟,38–100% B持续10分钟。
质谱数据经Maxquant软件处理,Maxquant分析考虑至少具有两个肽段的蛋白质,并根据特定的参数和说明搜索UniProt数据库。修饰方面,半胱氨酸的烷基化修饰被设置为固定修饰,而氧化(M)被考虑为可变修饰。设置蛋白质为胰蛋白酶的特异性剪切,最多允许两个漏切位点。片段质量容差设置为0.02 Da。为确保可靠的鉴定,要求蛋白质和肽段的最大假阳性发现率(FDR)均为1.0%。蛋白质的鉴定基于至少有一个唯一的肽段鉴定,而蛋白质的定量是通过计算每个蛋白质的唯一肽段的中位数来执行的。
每种方法产生了不同的蛋白质鉴定结果,如
图4A所示。总体而言,新方法鉴定的蛋白质数量最多(2975±52),其次是过滤辅助样品制备法(2964±102),最后是溶液法(2803±57)。每种方法的已鉴定肽段数量如
图4B所示。使用溶液法,鉴定了10931±16个独特的肽段。过滤辅助样品制备法和新方法分别鉴定了10981±48和10959±23个独特的肽段。平均而言,在溶液法中每个蛋白质匹配到3.90个独有肽段,在过滤辅助样品制备法中匹配到3.70个独有肽段,在新方法方法中匹配到3.68个独有肽段。这表明新方法表现出最高的蛋白质鉴定效率。
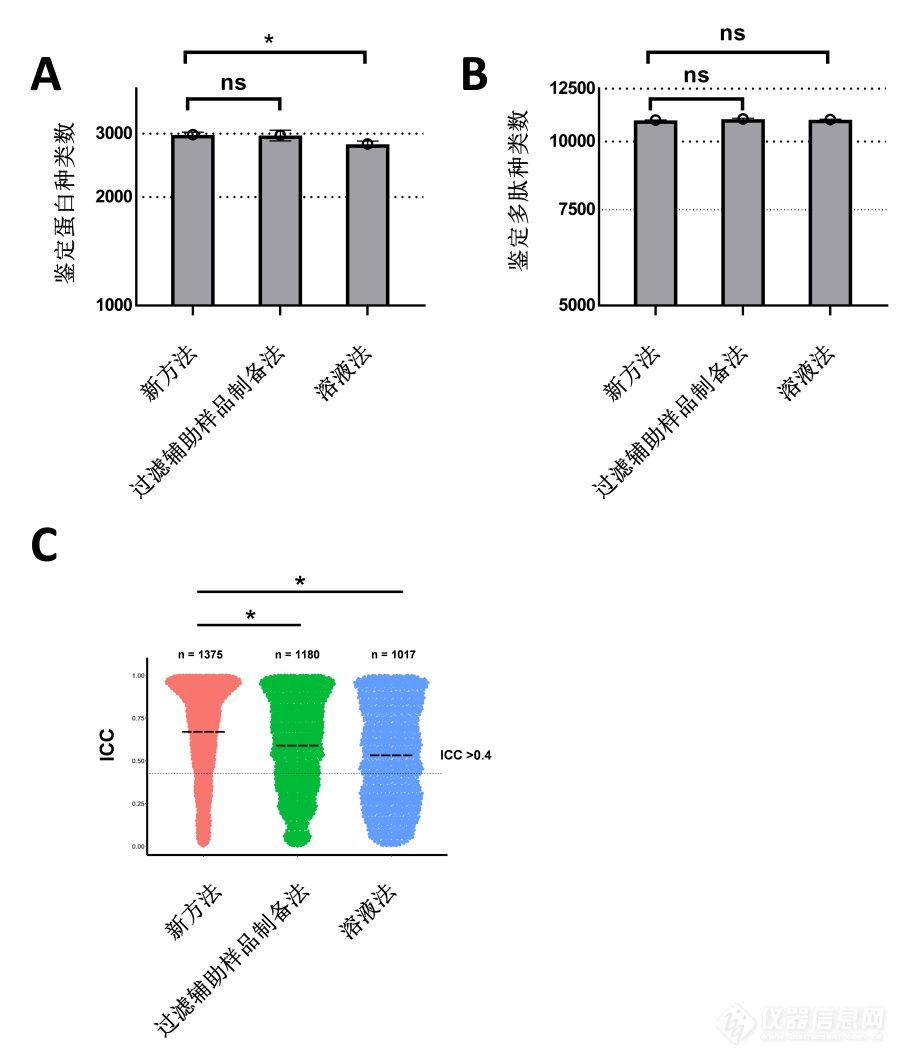
![]() 图 4 不同蛋白组学前处理方法在实际蛋白质组学分析中的应用。A:使用三种方法(溶液法、过滤辅助样品制备法和新方法)比较蛋白质鉴定结果;图B为每种消化方法鉴定的独特肽段,结果代表了三次生物学实验;图C为使用组内相关系数(ICC)评估无标签定量分析的可重复性;每种方法都有三次生物学重复。
图 4 不同蛋白组学前处理方法在实际蛋白质组学分析中的应用。A:使用三种方法(溶液法、过滤辅助样品制备法和新方法)比较蛋白质鉴定结果;图B为每种消化方法鉴定的独特肽段,结果代表了三次生物学实验;图C为使用组内相关系数(ICC)评估无标签定量分析的可重复性;每种方法都有三次生物学重复。随后,使用ICC评估无标签定量分析的可重复性(
图4C)。发现新方法显示出最佳的可重复性,平均ICC值为0.622,有1375个蛋白质的ICC值大于0.4。过滤辅助样品制备法表现出稍弱的可重复性,平均ICC值为0.533,有1180个蛋白质的ICC值大于0.4。相比之下,溶液法的可重复性最差,平均ICC值仅为0.477,有1017个蛋白质的ICC值大于0.4。
4.5 新方法有助于减少氨基酸残基的不利修饰
![]() 图 5 样品制备方法对氨基酸残基修饰的影响。A:比较三种样品制备方法(溶液法、过滤辅助样品制备法和新方法)引入的单氧化修饰;B:三种方法对氨基酸残基脱酰胺化修饰的影响,该实验作为生物学重复进行了三次。
图 5 样品制备方法对氨基酸残基修饰的影响。A:比较三种样品制备方法(溶液法、过滤辅助样品制备法和新方法)引入的单氧化修饰;B:三种方法对氨基酸残基脱酰胺化修饰的影响,该实验作为生物学重复进行了三次。氧化修饰和去酰胺修饰通常在天然蛋白质和多肽样本中被观察到,研究这些修饰对于理解蛋白质的固有应激至关重要。然而,样品预处理引起的人为修饰可能会对修饰蛋白组学分析带来挑战。为了评估在样品处理过程中引入的这些不利修饰对样品的影响,我们比较了三种方法:溶液法、过滤辅助样品制备法和新方法,关于它们对氧化和去酰胺修饰的影响。我们鉴定和定量了不同数量的单氧化和去酰胺修饰,但未检测到双氧化和三氧化修饰。在这三种方法中,新方法和溶液法显示出相对较低水平的单氧化修饰(
图5A)。此外,新方法还减少了非必要的去酰胺修饰(
图5B),鉴定了具有去酰胺修饰的蛋白质最少(473±8),其次是溶液法(485±23),最后是过滤辅助样品制备法(544±23)。考虑到有机溶剂中的羟基可以与酰胺键形成氢键,因此在溶液法和过滤辅助样品制备法中观察到较高水平的去酰胺修饰可能归因于在去盐过程中使用有机溶剂,而新方法更加高效的样品处理可能有助于减少人为的氧化修饰。总之,新方法有效减少了氨基酸残基的去酰胺和氧化修饰。
5? 结语
南开大学医学院实验室改进的蛋白组学样品前处理方法,利用超滤管将酶解缓冲液置换成碳酸氢铵水溶液,酶解后利用碳酸氢铵加热潮解的特性除去多肽溶液中的碳酸氢铵成分,最终得到纯净的多肽,可以方便地与
LC-MS直接衔接,进行质谱分析。与传统的方法相比,无样品损失,提高了蛋白质检测数目,同时操作简单快捷。值得注意的是,与传统的过滤辅助样品制备法相比,新方法在最小化氧化和去酰胺修饰方面表现出卓越的性能。这一创新的方法代表了蛋白质组学分析的重大进展,为质谱分析提供可靠和高效的结果。
虽然新方法和过滤辅助样品制备法在使用基于滤膜的蛋白质截断方法方面有相似之处,但它们之间的关键区别变得显而易见。过滤辅助样品制备法在酶解前需要进行更彻底的洗涤,通常需要使用碳酸氢铵水溶液进行5次洗涤,而过滤辅助样品制备法通常只需要进行2次洗涤
[20]。此外,新方法可以通过轻度加热实现原位样品脱盐和纯化方法的完美集成。相比之下,过滤辅助样品制备法通常需要额外的脱盐柱纯化步骤。与新方法和过滤辅助样品制备法不同,悬浮陷阱法(S-Trap)采用三维多孔材料来捕获蛋白质
[21]。由于其较大(亚微米级)的孔径,悬浮陷阱法滤膜的每个离心循环只需1分钟,实现了比本文中新方法和过滤辅助样品制备法更高的洗涤效率
[22]。然而,值得注意的是,与悬浮陷阱法相比,超滤管目前的成本较低,悬浮陷阱法在酶解后需要多次使用有机溶剂(如甲酸和乙腈)进行洗涤。悬浮陷阱法还涉及额外的脱盐柱纯化步骤,增加了实验的复杂性。总之,本文描述了一种独特的蛋白组学前处理方式,显著提高了高效样品制备的能力。
6? 数据可用性声明
本研究所有数据均包含在论文中。所有的蛋白质组学质谱数据已被存储在ProteomeXchange,并可通过访问编号PXD044209:
https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD044209 [23, 24]来获取。
参考文献(References):
[1]? Solari F A, Kollipara L, Sickmann A, et al. Two birds with one stone: parallel quantification of proteome and phosphoproteome using iTRAQ[M]//Proteomics in Systems Biology. Humana Press, New York, NY, 2016: 25-41.
[2]? Zhang G, Xue W, Dai J, et al. Quantitative proteomics analysis reveals proteins and pathways associated with anthocyanin accumulation in barley[J]. Food chemistry, 2019, 298: 124973.
[3]? Zhu Z, Chen T, Wang Z, et al. Integrated Proteomics and Metabolomics Link Acne to the Action Mechanisms of Cryptotanshinone Intervention[J]. Frontiers in pharmacology, 2021, 12: 700696.
[4]? Liu C, Si X, Yan S, et al. Development of the C12Im-Cl-assisted method for rapid sample preparation in proteomic application[J]. Analytical Methods, 2021, 13(6): 776-781.
[5]? Liu Q, Shi J, Sun J, et al. Graphene and graphene oxide sheets supported on silica as versatile and high‐performance adsorbents for solid‐phase extraction[J]. Angewandte Chemie, 2011, 123(26): 6035-6039.
[6]? Kecskemeti A, Bako J, Csarnovics I, et al. Development of an enzymatic reactor applying spontaneously adsorbed trypsin on the surface of a PDMS microfluidic device[J]. Analytical and bioanalytical chemistry, 2017, 409(14): 3573-3585.
[7]? Li L, Wu R, Yan G, et al. A novel method to isolate protein N-terminal peptides from proteome samples using sulfydryl tagging and gold-nanoparticle-based depletion[J]. Analytical and bioanalytical chemistry, 2016, 408(2): 441-448.
[8]? Shieh I F, Lee C Y, Shiea J. Eliminating the interferences from TRIS buffer and SDS in protein analysis by fused-droplet electrospray ionization mass spectrometry[J]. Journal of Proteome Research, 2005, 4(2): 606-612.
[9]? LaCava J, Molloy K R, Taylor M S, et al. Affinity proteomics to study endogenous protein complexes: pointers, pitfalls, preferences and perspectives[J]. Biotechniques, 2015, 58(3): 103-119.
[10]? Dupree E J, Jayathirtha M, Yorkey H, et al. A critical review of bottom-up proteomics: The good, the bad, and the future of this field[J]. Proteomes, 2020, 8(3): 14.
[11]? Duong V A, Park J M, Lee H. Review of three-dimensional liquid chromatography platforms for bottom-up proteomics[J]. International Journal of Molecular Sciences, 2020, 21(4): 1524.
[12]? 翟芳. 鸟枪法蛋白质组学质谱平台性能标准和参考数据集的建立[D]. 重庆大学, 2018.
[13]? Shen Y, Toli? N, Zhao R, et al. High-throughput proteomics using high-efficiency multiple-capillary liquid chromatography with on-line high-performance ESI FTICR mass spectrometry[J]. Analytical Chemistry, 2001, 73(13): 3011-3021.
[14]? Livesay E A, Tang K, Taylor B K, et al. Fully automated four-column capillary LC? MS system for maximizing throughput in proteomic analyses[J]. Analytical chemistry, 2008, 80(1): 294-302.
[15]? Ca?as B, Pi?eiro C, Calvo E, et al. Trends in sample preparation for classical and second generation proteomics[J]. Journal of chromatography A, 2007, 1153(1-2): 235-258.
[16]? Wheeler A R, Moon H, Bird C A, et al. Digital microfluidics with in-line sample purification for proteomics analyses with MALDI-MS[J]. Analytical chemistry, 2005, 77(2): 534-540.
[17]? Kim K H, Compton P D, Tran J C, et al. Online matrix removal platform for coupling gel-based separations to whole protein electrospray ionization mass spectrometry[J]. Journal of proteome research, 2015, 14(5): 2199-2206.
[18]? Wang C, Wu Z, Yuan J, et al. Simplified quantitative glycomics using the stable isotope label Girard’s reagent p by electrospray ionization mass spectrometry[J]. Journal of Proteome Research, 2014, 13(2): 372-384.
[19]? Righetti P G, Boschetti E, Lomas L, et al. Protein Equalizer? Technology: The quest for a “democratic proteome”[J]. Proteomics, 2006, 6(14): 3980-3992.
[20]? Homsi C, Rajan R E, Minati R, et al. A Rapid and Efficient Method for the Extraction of Histone Proteins[J]. Journal of Proteome Research, 2023, 22(8): 2765-2773.
[21]? Fu Q, Murray C I, Karpov O A, et al. Automated proteomic sample preparation: The key component for high throughput and quantitative mass spectrometry analysis[J]. Mass Spectrometry Reviews, 2023, 42(2): 873-886.
[22]? Duong V A, Lee H. Bottom-Up Proteomics: Advancements in Sample Preparation[J]. International Journal of Molecular Sciences, 2023, 24(6): 5350.
[23]? Ma J, Chen T, Wu S, et al. iProX: an integrated proteome resource[J]. Nucleic acids research, 2019, 47(D1): D1211-D1217.
[24]? Chen T, Ma J, Liu Y, et al. iProX in 2021: connecting proteomics data sharing with big data[J]. Nucleic Acids Research, 2022, 50(D1): D1522-D1527.
收稿日期:2023-10-27 修改日期: 基金项目:国家重点研发计划(32030083)。
作者简历:季学猛,硕士,实验师,研究方向为微生物学、蛋白组学。E-mail:jixuemeng@nankai.edu.cn。