获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 ken919 发表:
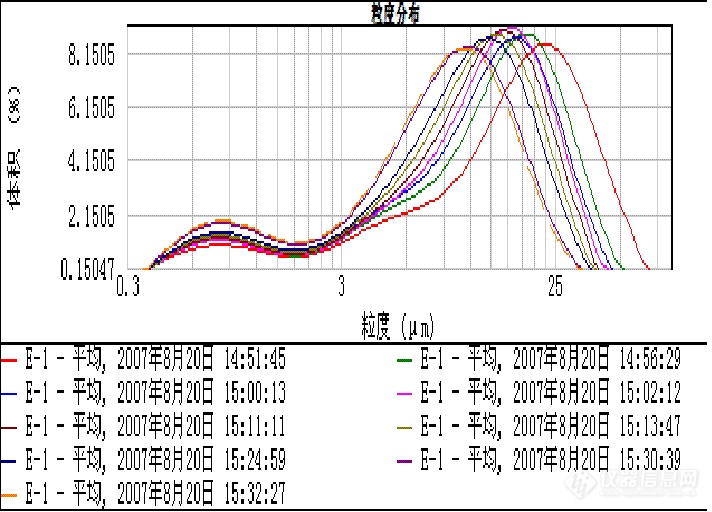
1.你可以将超声后的测量图谱叠加,如果小粒径越来越多,那么有可能样品被打碎,如果在一定范围内不会改变可能只是样品结块的被分散,
2.测量背景一般要求小于200,当然越小越好啦,这与你的系统的洁净度有关
3.一般用重量残差去衡量你系统的洁净程度
4.样品的物性决定光学参数,折光和吸光度对测量有一定影响,比如做OQ性能测试时,标准粒子的光学参数一定要准确输入才可能通过
5.分散剂的PH如果会影响样品的溶解度的话就有影响,气泡要避免,水系统可以先超声一分钟脱气才加入样品
原文由 ken919 发表:
不发意思,重量残差评估系统洁净的说法我也是听工程师说的/现在以前的贴子找到的说法是:残差是得出的粒度分布(即报告)按理论计算出的光强分布与实际检测器接受到的光强分布对比的差值,一定程度上可用来判断报告中光学参数的好坏,如果光学参数(折射率,吸收率)选的比较准确,则残差较小。值的大小由软件计算得出,与测试次数无关
上面的图是指一直超声后的测量读数还是重新加样的?要看超声后连续测量多次,每次间隔3分钟看看叠加图的变化 时间可以长一点
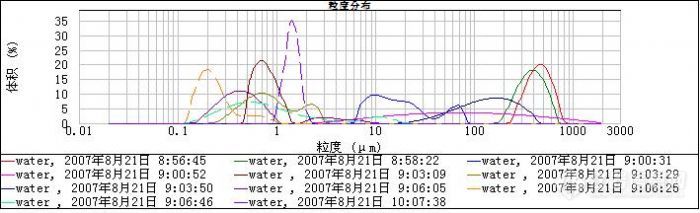
一般纯水分散剂是较洁净的,但会有一些微粒在里面但应作为背景可以扣除,所以对结果影响不大.
有时样品池有气泡也会这样,光路系统不干净也会这样,你主要分析背景就可以了吧,我记得ME一程师说过看原始数据图就是有背景和样品信号的那个图