后续将有专属客服与您沟通!
关注微信公众号查看留言进度 接收留言处理通知
0
ID:chqiu111111
行业:其他
积分:0升级还需100积分
声望:0升级还需100声望
注册时间:0000-00-00
最后登录时间:0000-00-00
请确认联系方式
请输入您的联系方式
提交留言即视为您同意遵守 《服务协议》和 《隐私权政策》
ID:lingshike
ID:huaibeijiayuan
ID:tianhaikuo
ID:laibuxyf
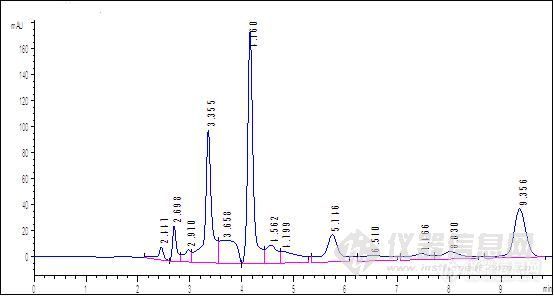
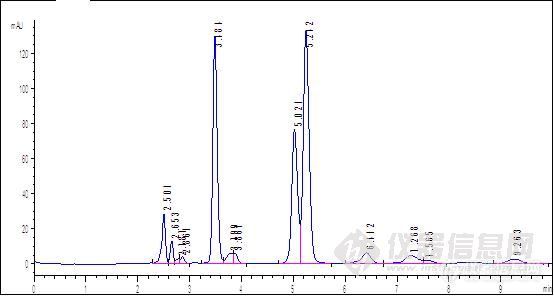
原文由 huaibeijiayuan(huaibeijiayuan) 发表:会不会是样品残留呢,PH调整后,强保留物质被洗脱出来了
原文由 tianhaikuo(tianhaikuo) 发表:流动相ph不同,其极性就不同,对相同物质的洗脱能力或保留能力也不同。
ID:YJYQXCYS
原文由 东风恶(luoleqc) 发表:建议结合您的合成工艺,参考相关文献,对起始原辅料进行相应的分析,可以用DAD检测器扫描光谱图,还有您的中间体。根据您的合成工艺初步判断可能产生的物质来慢慢摸索。
ID:yangxy12345
ID:zxhcnf