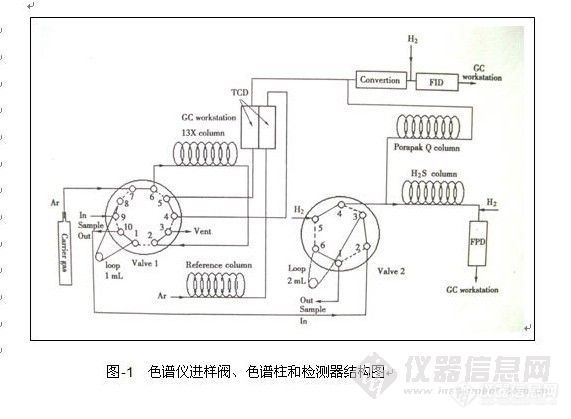
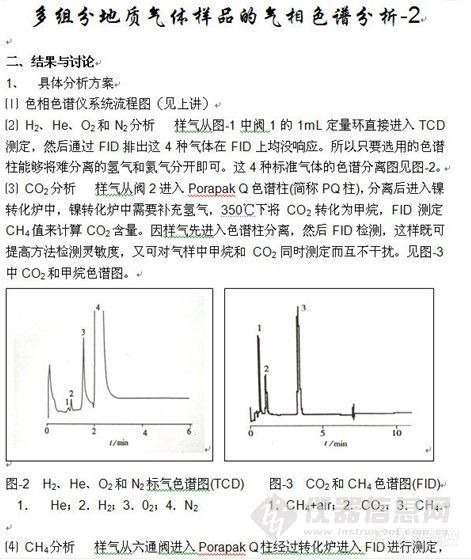
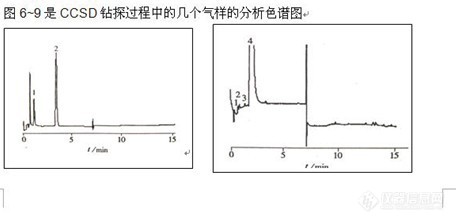
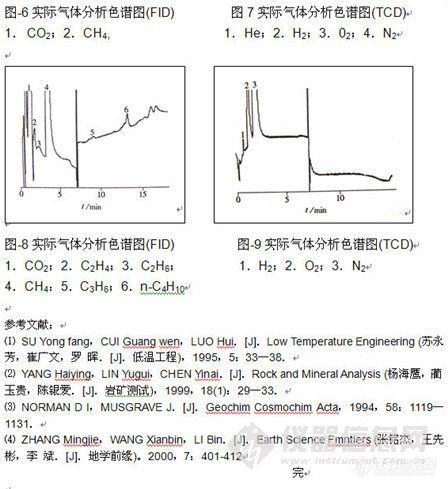
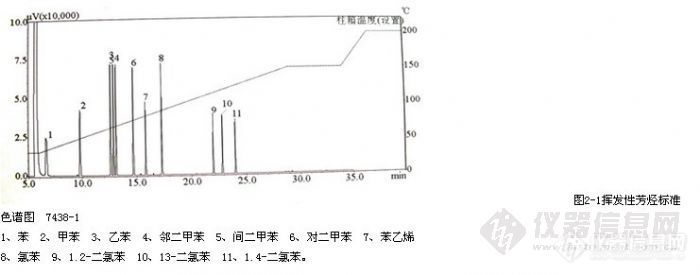
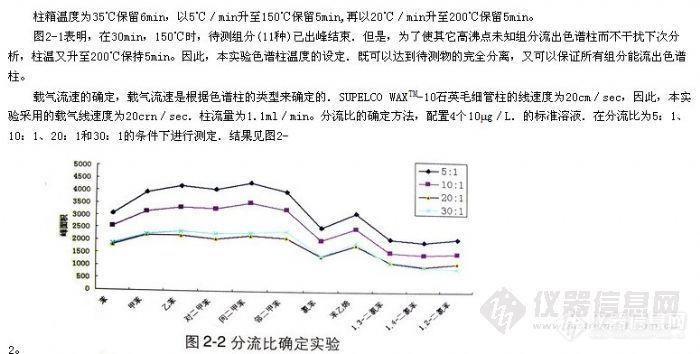
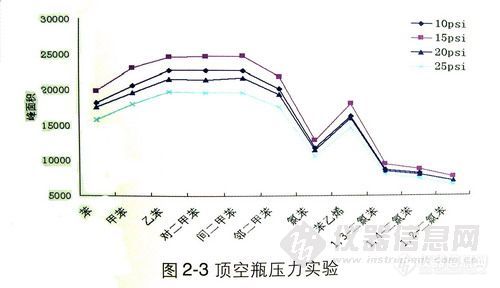
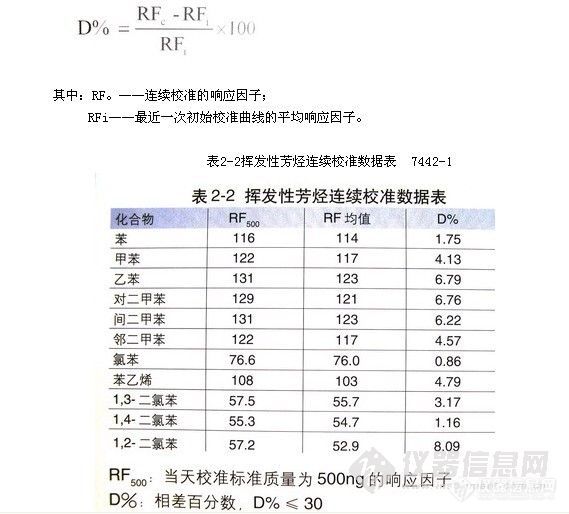
顶空气相色谱系列讲座(93) 顶空固相微萃取-
气相色谱-质谱法测定
纺织品中防虫蛀剂的残留
前言
采用顶空固相微萃取技术和
气相色谱-质谱联用技术对纺织品中的挥发性防虫蛀剂的残留进行了测定。该方法对挥发性二氯苯和萘的检测限量为1μg/kg,回收率为83.6%~l15.2%,相对标准偏差为8.1%~9.8%。
随着社会的进步和全球环保意识的提高,人们对服装的要求也逐渐从满足于传统的防皱免烫、防虫蛀、抗菌除臭等功能转为倡导清洁生产和绿色消费,开始从环境生态的角度去认识纺织品中有害物质的潜在危害[1~3]。目前,发达国家已制定了许多环保贸易法令,通过产品技术限制和质量标准规范的形式,对纺织品生产过程中的各个阶段给环境和人体造成的影响做了严格规定,要求服装中不得含有对人体造成伤害的有毒有害物质,不得存在潜在的、可能对人体或其他物体造成伤害的因素[4]。检验纺织品中的农药和其他有害物质对于保护消费者权益、提高纺织品的生态标准有着积极的意义[5]。高档织物中防虫蛀剂残留的安全限量始终是困扰生态纺织品标签认证的问题之一,有关农药及其他有害物质在毛织物中蓄积水平的确认和最终代谢产物的鉴定,是评价纤维中外来分子毒性和归宿的基本前提[6]。目前已见于文献的各种织物的功能增效剂、驱虫剂和防霉剂残留检测多为借助
气相色谱(GC)和高效
液相色谱(HPLC)对防虫蛀剂中的有效成分进行定性定量分析[7,8]。本文采用顶空固相微萃 (HS-SPME) 技术,通过
气相色谱与质谱联用技术 (GC-MS) 的定性定量分析,探讨适用于纺织品中防虫蛀剂残留的测定方法。
本文是苏州出入境检验检疫局陈军同志的获奖项目,现介绍陈军撰写的获得国家出入境检验检疫局科技兴检三等奖中的部分内容,供网友们参考
1、实验部分
1-1试剂
分析纯试剂:甲醇重蒸后使用、氯化钠。缓冲溶液:A液(0.2mol/L硼酸+ 0.1moL/L柠檬酸) +B液(0.1mol/L磷酸钠),pH为2.2,V(A):V(B) = 4:1。
标准物质:l,4-二氯苯 (p-DCB)、l,3-二氯苯 (m-DCB)、l,2-二氯苯 (o-DCB) 和萘,均由Chem Service提供。
标准溶液的制备:取适量标准物质,用甲醇配成1mg/L的标准储备液,于-4℃下保存。使用时根据实际需要稀释、混合后制成相应的标准工作液。
标准贴衬:毛标准贴衬 (ISO 105/F01-1982)。
水溶液用超纯水配制,玻璃容器和磁力搅拌器用超纯水净化。
1-2仪器设备
HP-6890 GC
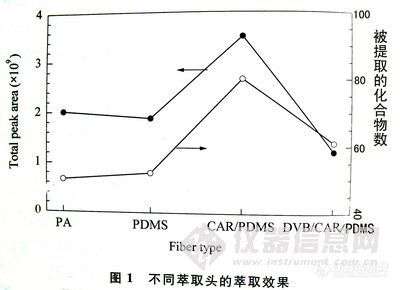
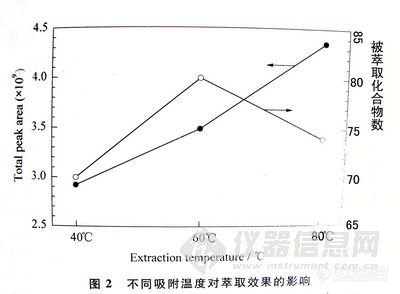
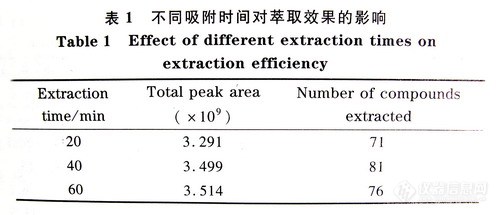
气相色谱仪和5972-MSD质谱仪。熔融石英毛细管柱:HP-5 ,30m×0.25mm i.d.×0.25μm。柱头压:70kPa (He)。进样口温度:230℃。检测器温度:300℃。进样体积:lμL,不分流进样方式。程序升温:初温40℃,以25℃/min的速率升至l80℃,停留lmin,再以2℃/min的速率升至205℃,停留3min,然后以10℃/min的速率升至260℃,停留12min,结束。固相微萃取器:GC专用;固相微萃取纤维:l00μm聚二甲基硅氧烷(polydimethylsiloxane,PDMS Supelco公司).。Branson超声波机。
1-3分析步骤
用20cm×2Ocm聚乙烯塑料袋到现场直接取样,密封后带回实验室供测定用 (样品应尽快分析,在冰箱内保存一般不要超过24h)。分析前在标准温湿度条件下将样品粉碎,准确称取0。0.2g样品,置于4mL样品瓶内,加入3mL含300g/L NaC1及5% (体积分数) 甲醇的缓冲溶液(pH2.2),用衬有聚四氟乙烯 (PTFE) 隔垫的瓶塞封口。于(50±1)℃下超声处理90min,待样品充分浸润、纤维完全膨胀后即可进行SPME操作。操作时,将纤维头置于样品液面上方(约1mm),开动磁力搅拌器,加热至(40±1)℃,萃取5min。完成萃取后,再将SPME针管插入GC进样口,于230℃下热脱附3min。每个样品测试均进行3次平行实验(精密度测定除外),测定结果取平均值。
2、结果与讨论
2-1质谱鉴定与保留时间定性
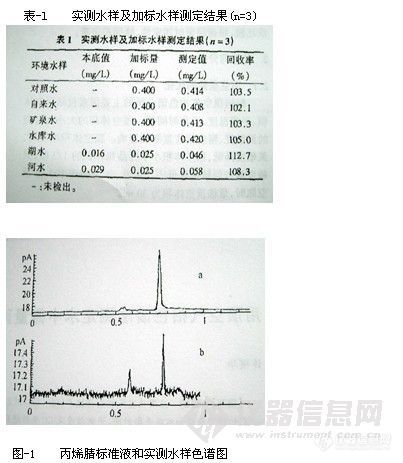
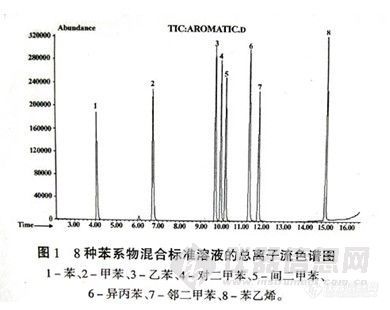
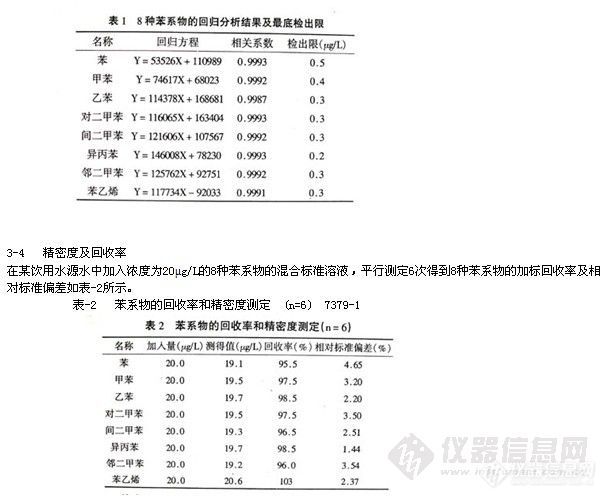
吸取适量标准储备液,用含300g/L NaCl的甲醇-缓冲溶液 (体积比为5:95) 配制成10μg/L的挥发性二氯苯 (p-DCB,m-DCB和o-DCB) 及50μg/L萘的混合标准溶液,共两组,分别用100μm的PDMS对它们进行SPME操作,在选定的仪器条件下得到顶空固相微萃取的
气相色谱一质谱定性结果 (见表-1)。所有目标化合物在选定的GC-MS条件下均有良好的分离度,出峰时间间隔足以在选择离子方式(SIM)下定量设定多个时间窗口,以使上述目标化合物能同时进行SIM测定。DCB多见于衣物的卫生除虫制剂中,主要成分是p-DCB,,GC-MS能够检测出它的两种异构体m-DCB和o-DCB,其碎片离子m/z l46,l11,75和p-DCB非常相似,因其检出量与p-DCB相比不够显著,故本研究未专门讨论二氯苯异构体的鉴别,定量分析也仅考虑衣物中防虫蛀制剂的主要成分p-DCB的残留。
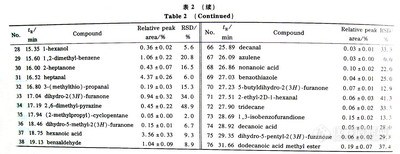
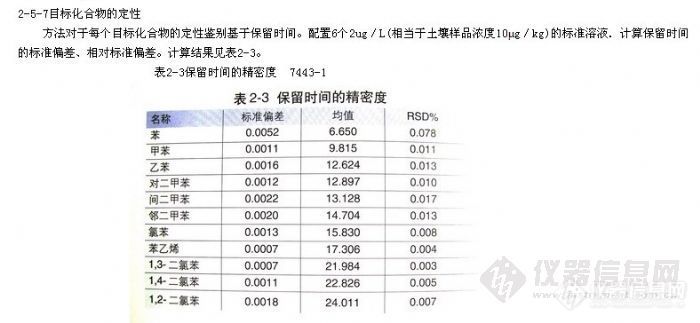
表-1 目标化合物的保留时间及有关参数(7365-1)
![]()
![]()
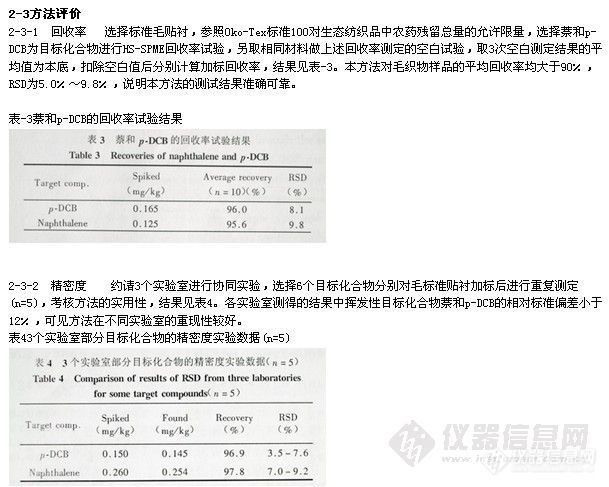
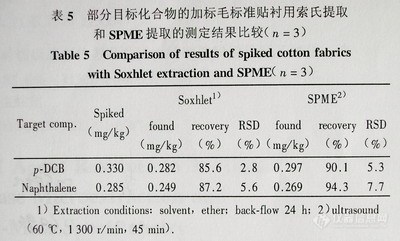
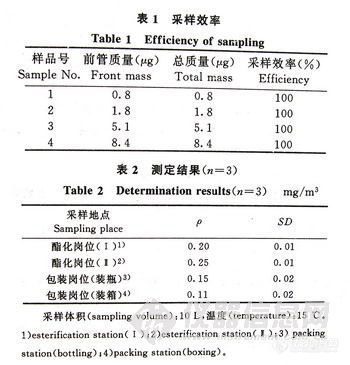
2-3-3 索氏提取和SPME提取的比较 取相同加标样品,分别以索氏提取和SPME提取两种方法进行目标化合物加标回收率的测定。结果发现,低含量的挥发性目标化合物的SPME提取率优于索氏抽提,3次重复测定的结果均高于索氏抽提的测定值 (见表5)。
表5部分目标化合物的加标毛标准贴衬用索氏提取和SPME提取的测定结果比较(n=3)
![]()
1)Extractionconditions:solvent,ether;back.fl0w 24h;
2)ullras0und(60℃,1300r/min,45min)
2-4 实样分析
在实验中选用毛纱及部分市售印花织物样品进行分析,按照Oko-Tex标准100和MST标签的限量规定,在部分织物样品中检出少量的萘,质量比为0.07mg/kg~0.16mg/kg。在所有样品中,均未测出二氯苯。
3、结论
本研究参照国内外有关生态纺织品的要求,将顶空固相微萃取技术应用于纺织品有害物质检测领域。所确立的甲醇-硼酸-柠檬酸-磷酸钠缓冲体系有效地解决了织物纤维无溶剂化萃取的基质效应问题。对选定的目标化合物进行分析,得到了满意的结果。该方法对挥发性二氯苯和萘的检测限量 (LOQ) 为1μg/kg,回收率为83.6%~115.2%,相对标准偏差为8.1%~9.8%。
参考文献:
1、 Schlichter S,Kuschel A,Melliand Textilber,1995,76(4):E49
2、 Kermer W D.Melliand Textilber,1995,76(2):El4
3、 Pfaendr F K,Jones R B,Stevens A A,et a1.Environ Sci Technol,1998,42(4):438
4、 OKO-Tex Standard 100 (2000ed) .Vienna (Austria) :International Association for Research and Testing in the Field Ecology,2000.2
5、 Health D F,Vandeker M.Brit J Industr Med,1984,41:269
6、 Peters R H.Textile Chemistry,1995,111(18):581
7、 International Wool Secretariat.TM27:Testing Methods for Pure Wool Products. Melbourne (Australia):Pawliszyn Pub,1990.20
8、 Theresa A P,Rhonda P W.Colourage,1995,56(7):55