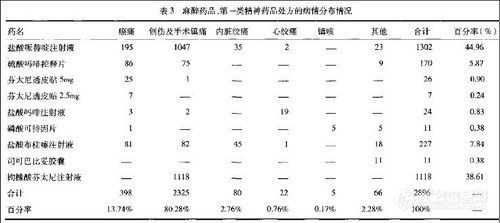
兴奋剂类分析方法系列讲座(23)2种液相色谱分离模式测定半夏露糖浆中麻黄碱和伪麻黄碱 用2种高效
液相色谱分离模式分析测定半夏露糖浆中
麻黄碱、伪麻黄碱成分
摘 要 目的:比较采用反相键合相
液相色谱法和离子交换
液相色谱法分析测定中成药半夏露糖浆中麻黄碱、伪麻黄碱成分。
方法:色谱条件1以AgilentZORBAXExtendC18柱为色谱柱,流动相为0.2%磷酸溶液-乙腈(95:5),流速1.0mL·min-1,检测波长206nm;色谱条件2则以SpherisorbSCX柱为色谱柱,流动相为75mmol·L-1磷酸盐溶液(pH7.0)-乙腈(85:15),其余同色谱条件1。供试品溶液以蒸馏法获得。
结果:采用2种
液相色谱法,供试品中麻黄碱和伪麻黄碱成分均可得到完全分离,盐酸麻黄碱、盐酸伪麻黄碱进样量分别在0.05~0.5μg及0.03~0.3μg与峰面积呈良好线性关系。2种色谱方法所测得结果一致。
结论:2种
液相色谱法均可用于测定半夏露糖浆中麻黄碱、伪麻黄碱的含量,另采用离子交换色谱法测定可较直观地得到麻黄中包含生物碱在内的碱性成分的指纹信息。
麻黄碱、伪麻黄碱盐酸盐是国家控制的能制备毒品的原料药,都具有拟肾上腺素作用和对中枢神经系统兴奋作用,运动员要服用这种止咳糖浆就会引来麻烦。本文由姜舜尧2 田颂九1 刘琼3 (1.中国药品生物制品检定所; 2.浙江省药品监督管理局; 3.浙江英特药业有限责任公司) 用二种实验模式来分离检测半夏露糖浆获得可靠结果。现全文介绍如下。
前 言
半夏露糖浆系一常用止咳化痰类中成药制剂,由生半夏、麻黄、枇杷叶、远志、款冬花、桔梗、陈皮、甘草、薄荷油9味中药制成。处方中主药麻黄含多种结构相似的麻黄碱类生物碱,如麻黄碱、伪麻黄碱及少量的去甲麻黄碱、去甲伪麻黄碱、N-甲基麻黄碱、N-甲基伪麻黄碱等,这类成分都具有拟肾上腺素作用和对中枢神经系统兴奋作用[1,2],在本制剂中有必要对这类成分作定量控制。本文运用2种高效
液相色谱分离模式(反相
液相色谱法和离子交换
液相色谱法)对样品中所含有的麻黄碱、伪麻黄碱成分进行了分析测定。
实 验
1 仪器及试药
Agilent1100高效
液相色谱系统:G1311A四元泵,G1322A脱气机,G1313A 自动进样器,G1316A柱温箱,G1315B二极管阵列检测器,A08.03版Agilent化学工作站。对照品盐酸麻黄碱、盐酸伪麻黄碱及麻黄对照药材均由中国药品生物制品检定所提供。半夏露糖浆(规格:100 mL 相当于含生药13.38g)3批,由药厂提供。乙腈为色谱纯,磷酸、磷酸二氢钠、磷酸氢二钠、氢氧化钠、氯化钠均为分析纯,重蒸馏水。
2 色谱条件
条件1:色谱柱为Agilent ZORBAXExtendC18柱(250mm×4.6mm,5μm),流动相为0.2%磷酸溶液-乙腈(95:5),流速1.0mL·min-1,柱温25℃,检测波长206nm。
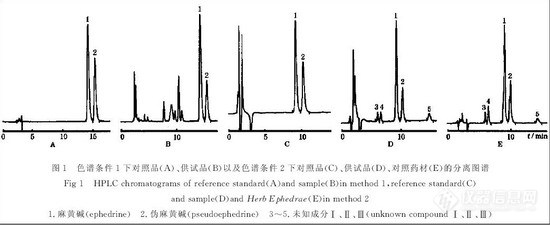
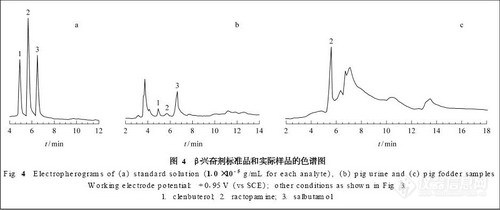
条件2:色谱柱为SpherisorbSCX (150mm×4.6mm,5μm),流动相为75mmol·L-1磷酸盐溶液 (75mmol·L-1磷酸氢二钠溶液用75mmol·L-1磷酸二氢钠溶液调pH 至7.0)-乙腈(85:15),其余同条件1。在两色谱条件下供试品及在色谱条件2下麻黄对照药材的色谱分离结果如图1所示。
![]()
3 溶液的制备
3.1 对照品溶液
取对照品盐酸麻黄碱、盐酸伪麻黄碱适量,分别加水稀释制成9.936μg·mL-1的盐酸麻黄碱对照品溶液和6.24μg·mL-1的盐酸伪麻黄碱对照品溶液。
3.2 供试品溶液
精密称取半夏露糖浆约25g,置蒸馏瓶中,加水10mL,加氯化钠7.5g和20%氢氧化钠溶液100mL,蒸馏,用预先盛有0.5mol·L-1盐酸溶液5mL的100mL量瓶收集蒸馏液近95mL,加水至刻度,摇匀,得供试品溶液。
3.3 麻黄对照药材溶液
取麻黄药材0.5g,按供试品溶液制备方法制得麻黄对照药材溶液。
4 分析方法论证
4.1 线性范围
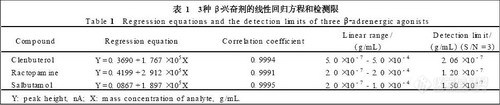
取盐酸麻黄碱及盐酸伪麻黄碱2种对照品溶液,分别于两色谱条件下分别进样5,10,20,30,40,50μL,对所测得的峰面积(Y)和各对照品进样量 (X) 作回归处理,结果表明,麻黄碱、伪麻黄碱进样量分别在0.05~0.5μg及0.031~0.31μg呈良好线性关系,相关系数r均大于0.9998。
4.2 进样精密度和重复性
取盐酸麻黄碱和盐酸伪麻黄碱2种对照品溶液,分别于两色谱条件下重复进样8次,进样精密度RSD (n=8)在0.4%~0.8%之间;取同批半夏露糖浆6份,按上述方法制得供试品溶液,在色谱条件1下测定麻黄碱和伪麻黄碱含量,方法重复性RSD (n=6)分别为0.6%和0.5%,在色谱条件2下测定,RSD(n=6)分别为1.0%和1.8%。
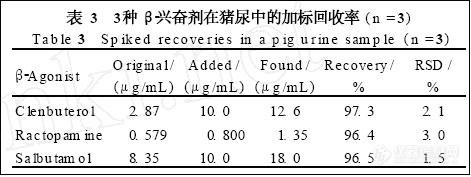
4.3 回收率试验
精密称取已知含量的半夏露糖浆(含盐酸麻黄碱和盐酸伪麻黄碱量分别为0.0382,0.015mg·g-1)约12.5g各6份,分别加入盐酸麻黄碱对照品0.4968mg和盐酸伪麻黄碱0.187mg,按上述方法提取制备,于色谱条件1下测定,计算麻黄碱和伪麻黄碱的平均回收率 (n=6)分别为100.5% (RSD=1.2%) 和102.5% (RSD=2.2%),于色谱条件2下测定,则2成分平均回收率 (n=6) 分别为101.5% (RSD=1.7%) 和101.8% (RSD=2.5%)。
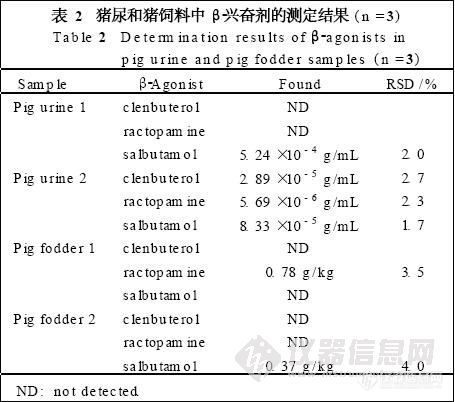
5 样品测定结果
在色谱条件1下,测得3批样品含麻黄碱量分别为0.038,0.043,0.044mg·g-1,含伪麻黄碱量分别为0.015,0.017,0.017mg·g-1;在色谱条件2下测得结果与色谱条件1下所得结果一致。
6 离子交换色谱条件下供试品的色谱分离行为采用SCX柱以离子交换色谱法分析供试品中
生物碱成分,流动相pH 及其离子浓度是影响带正电
离子色谱行为的重要因素[3~7]。
6.1 流动相pH 对色谱分离的影响
取75mmol·L-1的磷酸溶液、磷酸二氢钠溶液、磷酸氢二钠溶液,互相调节配制成不同pH 的75mmol·L-1磷酸盐溶液,以磷酸盐溶液-乙腈 (85:15) 作为流动相,各自测定峰1~5 (图1) 的保留因子。实验发现,流动相pH在1.8~3.0范围变化对峰1~5的保留因子均影响很大,随pH 增大,各成分保留因子减小,而pH4.0以上峰1~5的保留因子则基本不受pH变化影响。
6.2 流动相离子浓度对色谱分离的影响
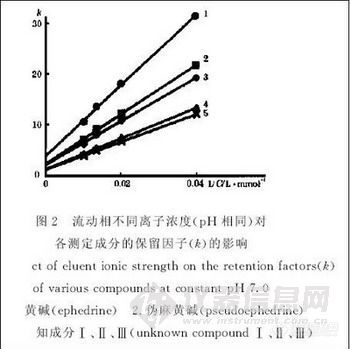
配制浓度分别为20、50、75、100mmol·L-1的磷酸氢二钠溶液及磷酸二氢钠溶液,互相调节配制成pH 为7.0的不同浓度的磷酸盐溶液,以磷酸盐溶液-乙腈 (85:15)作为流动相,各自测定峰1~5的保留因子,以磷酸盐浓度的倒数与测定物的保留因子变化作图(图2)。可看出,流动相其离子浓度倒数与各测定物的保留因子的变化均可成一斜率为正值的直线,根据离子交换色谱原理,表明此时各测定物的色谱分离均存在着离子交换保留机理[3~6]。
![]()
7 供试品中麻黄所含碱性成分的色谱指纹识别
从图1中(D)、(E)可看出,半夏露糖浆供试品色谱中的峰1~5在麻黄对照药材色谱中均能一一对应,另以处方中去麻黄的其余8味药按半夏露糖浆制备工艺、供试品溶液制备方法制得缺麻黄空白对照溶液,进样分析可证实峰1~5均为麻黄药味所特有;图2表明,峰1~5在色谱柱上存在着离子交换作用,结合在流动相pH 变化情况下这类成分的色谱行为,可判断均为带正电荷成分;另外,用二极管阵列扫描发现峰1~5的紫外光谱类同。上述实验可判断峰3~5为麻黄中的碱性成分,且推测可能为麻黄碱类生物碱成分。
8 讨论
2种
液相分离模式均可用于半夏露糖浆中麻黄碱、伪麻黄碱成分的定量测定。另外,采用SCX柱以离子交换色谱法分离,供试品中的酸性成分和中性成分不被保留[7],生物碱等碱性成分可获得较好的色谱分离,故在其色谱中可较直观地得到麻黄中含生物碱在内的碱性成分的指纹信息。
![]()