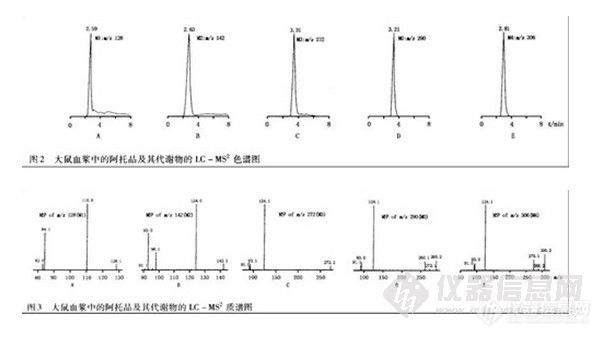
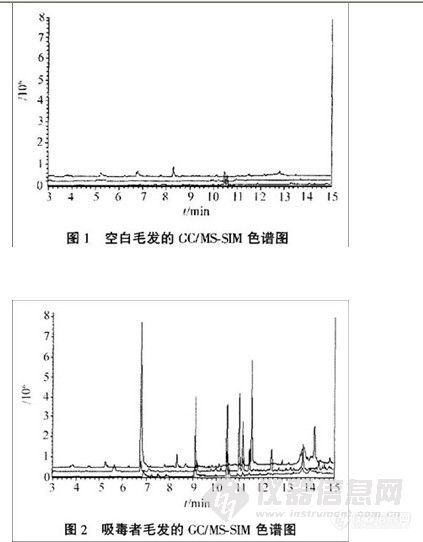
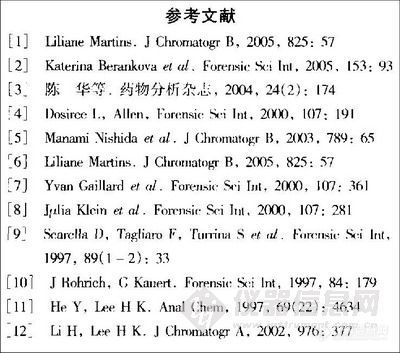
兴奋剂类分析方法系列讲座(34)动态液相微萃取CC/MS-SIM方法检验毛发中的苯丙胺类毒品 动态
液相微萃取GC/MS-SIM方法
检验毛发中的苯丙胺类毒品
摘 要:建立了动态
液相微萃取GC/MS-SIM方法检测毛发中4种苯丙胺类毒品的方法。毛发样品首先用1 mol/L Na0H溶液消解,然后用50μL.氯仿涡旋提取1min,离心后用注射器直接抽取有机相,提取液进行CC/MS-SIM方法检测。毛发样品的测出限(S/N=3)分别为苯丙胺l ng/mg,甲基苯丙胺、3.4-(亚甲二氧基)苯丙胺、3.4(亚甲二氧基)-甲基苯丙胺500 pg/mg。在毛发中添加上述4种苯丙胺毒品的质量分数为5 ng/mg时,5次测定的RSD分别为苯丙胺8.3%,甲基苯丙胺8.2%,3、⒋(亚甲二氧基)苯丙胺2.0%,3、4(亚甲二氧基)甲基苯丙胺2.7%。该方法可用于毛发中低含量苯丙胺类毒品的分析。
本文由中国人民公安大学刑科技系的朱 丹、孟品佳*、路春清老师们共同完成,现全文介绍如下。
前 言
苯丙胺类毒品是一类具有强烈兴奋作用、食欲抑制作用以及温和致幻作用的物质,属于违禁毒品。在国际和国内均受到严格控制。滥用MDMA(摇头丸)、甲基苯丙胺(冰毒)兴奋剂导致精神障碍和行为异常、引发许多社会问题。因此,很多的国内外学者关注苯丙胺类毒品的研究。判断一个人是否吸食苯丙胺类毒品,通常是通过毛发和体液来检验。由于毛发与体
液相比,具有易获取、稳定、易保存及不易作假等优点。所以,国外已经有很多通过毛发检验苯丙胺类毒品的相关报道。
目前检测毛发中苯丙胺类毒品的常规方法是首先把毛发消解,然后经过固相萃取[1,2]、
液相萃取[3]、超临界流体萃取[4]等萃取技术对消解液进行萃取,然后把萃取液吹干后进行衍生化[5]:最后采用
气相色谱法(GC),
气质联用(CC/MS)[6],高效
液相色谱法(HPLC)[7]、放射免疫法(RIA)[8]和高效毛细管电泳[9]等作为检测手段进行检测。 在相关的文章所做的检测里,毛发的苯丙胺质量分数在0.96~15.8 ng/mg或者0.5~O.9 ng/mg(甲基苯丙胺的代谢物),甲基苯丙胺在0.6~56.5 ng/mg,MDA 在0.04~5.0 ng/mg, MDMA 在 0.05~6.4ng/mg [10],MDE在0.8~17 ngr/mg [10]。在许多案件里,苯丙胺和甲基苯丙胺都是要检验的毒品。
本文主要考查了毛发中4种苯丙胺类毒品,苯丙胺(AM)、甲基苯丙胺(MAM)、3、4-(亚甲二氧基)苯丙胺(MDA)、3、4-(亚甲二氧基)-甲基苯丙胺(MDMA)的消解、萃取、检测方法。在萃取过程中采用了动态
液相微萃取[11,12]的方法,在GC/MS检测方法中采用了选择离子检测(SIM)方法。与全扫描(SCAN)方式比较,检出限可提高数10倍。采用GC/MS-SIM方法,不经衍生化,可使得苯丙胺的毛发样品的测出限(S/N=3)达到1 ng/mg,甲基苯丙胺、3、4-(亚甲二氧皋)苯丙胺、3、4-(亚甲二氧基)-甲基苯丙胺的毛发样品的测出限(S/N=3)达到500 pg/mg。本文所建立的方法灵敏、快速、可靠、简便,可以应用于缉毒、戒毒的实际工作中。
实 验
1 实验部分
1.1 仪器与试剂
岛津5050
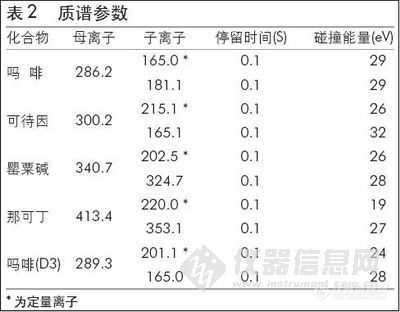
气相色谱仪,配有质谱检测器;色谱柱键合DB-1色谱柱,30 m×0.32 mm i.d.,1μm;进样器温度250℃,柱箱起始温度100℃,保持2 min,然后以10℃/min升至220℃,保持2min;氦作载气,流速2.0 mL/min,不分流;质谱检测器温度:280℃,选择离子为:(m/z) 58、91、136,相对电压0.1 eV。
苯丙胺盐酸盐(AM)、甲基苯丙胺硫酸盐(MAM)、3、⒋(亚甲二氧基)苯丙胺盐酸盐(MD`)购于国家麻醉品实验室;3、4(亚甲二氧基)-甲基苯丙胺盐酸盐(MDMA),购于公安部物证鉴定中心;2甲基苯乙胺购于ACROS ORGANICS公司;所用试剂均为分析纯。
标准溶液的配制:准确称取AM 2.7g、MAM 1.2g、MDA 1.2g、MDMA 2.2g,用甲醇溶液配制成1 mg/mL的标准溶液(以游离碱计)。
内标溶液的配制:准确称取2-甲基苯乙胺1mg,用甲醇溶液配制成l0μg/mL的2-甲基苯乙胺的内标溶液。
1.2 实验方法
取空白头发(非吸毒人的头发),用二氯甲烷振荡洗涤2次,晾干后剪成长约1 mm。取约20mg,准确称定,加人含有内标溶液的苯丙胺类4种毒品的混样(其中内标溶液的质量浓度为10 μg/mL),加人1 mol/L的Na0H溶液1 mL,15℃下水浴加热30min,取出,加人少许KCl(必需使溶液达到过饱和),然后加人氯仿50μL,涡旋提取l min后离心1 min,取上层液直接进样,GC/MS-SlM方法检测。
2 结果与讨论
2.1 检测离子的选择
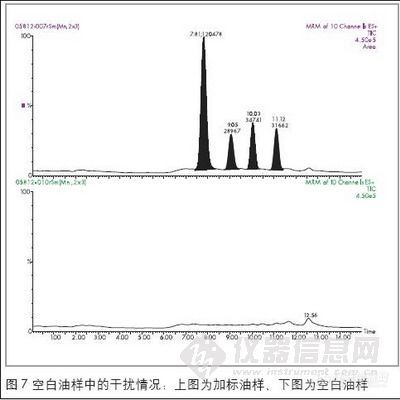
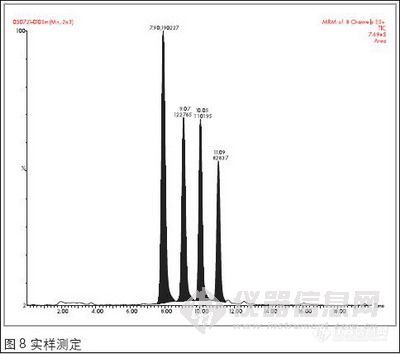
为了能满足检测灵敏度的要求,并且能够减少其它物质的干扰,故选择响应最高的离子进行检测:因MAM和MDMA都有m/z 58的离子且丰度较高,所以选择rn/z 58作为MAM和MDMA的定量离子,囚AM和2-甲基苯乙胺都有rn/z 91的离子,且丰度较高,所以选择m/z 91 作为MAM和2-甲基苯乙胺的定量离子,而选择rn/z 136为MDA的定量离子、在GC/MS-SCAN条件下。测定标准品得出各种苯丙胺类毒品的保留时间,共中,AM为5.83 min,MAM为6.78 min,2-甲基苯乙胺为7.13min,MDA为10.99 min,MDMA为l 1.59min。
2.2 线性范围及检出限
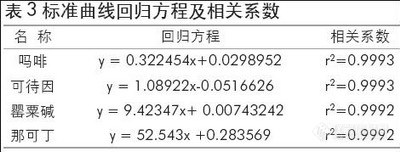
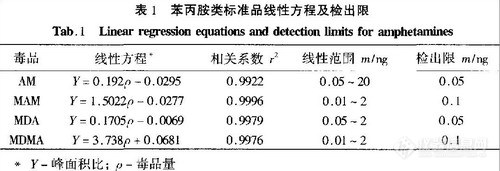
用质量浓度分别为0.05、0.2、2、4、10、20μg/mL的AM标准品(均含内标10μg/mL),和质量浓度分别为0.01、0.05、0.1、0.2、1、2 . μg/mL的MAM、MDA、MDMA的标准品(均含内标10μg/mL),在下述GC/MS-SIM条件下分别进1μL以进样质量浓度为横坐标,以苯丙胺类毒品的峰面积和内标二甲基苯乙胺的峰面积的比值为纵坐标获得苯丙胺类毒品的线性方程。从表1中可以看出此方法线性良好,方法可行。
![]()
2.3 最佳萃取条件的选择
2.3.1 涡旋时间的选择
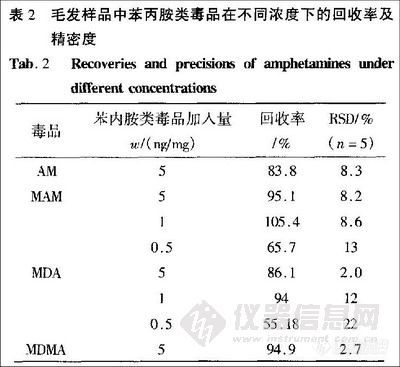
取清洗后剪碎毛发约20mg,准确称定,加人含有内标溶液的苯丙胺类4种毒品的混样(其中内标溶液的质量浓度为l0μg/mL,4种苯丙胺类毒品的质量浓度都为2μg/mL)50 μL,然后加人1 mol/L的NaOH溶液1mL,75℃下水浴加热30 min,取出,加人CHCl3 50μL,分别旋涡20 s,1.2 min,然后离心1 min,取下层液进样,GC/MS-SIM方法检测。以每种毒品的峰面积和内标2-甲基苯乙胺的峰面积之比为参数,比较提取率。根据实验结果,涡旋时间l min时 提取率较高。涡旋20 s时萃取进行不完全,当涡旋2min时,由于涡旋时间过长导致苯丙胺类毒品的挥发,使提取率降低,所以涡旋1 min较为适宜。
2.3.2 加盐的作用
头发的消解过程如2.3.l,消解后的溶液不加人KCl和加人足以使溶液过饱和的少许ΚCl,然后加人CHCl3 50 μL,涡旋1 min后离心1min, 取下层液进样,GC/MS-SIM方法检测,以每种毒品的峰面积和内标2-甲基苯乙胺的峰面积之比参数比较提取率。由于盐具有与有机相竞争水分子的作用,加盐可提高提取率。
2.3.3 提取溶剂用量
头发的消解过程如2.3.1,消解液加人少许KCl后(使溶液达到过饱和),分别加人CHCl3溶液20、40、50、100、200 μL,旋涡、离心,取下层液进样,GC/MS-SIM方法检测,实验表明CHCl3用量越小,提取率越高。因为该方法是取有机相直接进样分析,因此有机溶剂用量越少,有机相中待测物浓度越大,检测的灵敏度越高。但有机相体积过小,不易抽取液体,故选择50 μL为有机相提取体积。