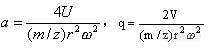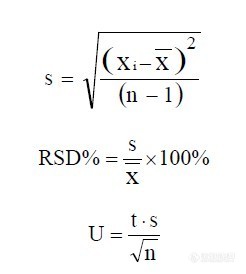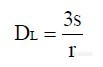[1106] 维权声明:本文为lidonglei原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现的,均属侵权违法行为,我们将追究法律责任。
用前言做个说明,具体内容请看附件,技术问题欢迎讨论。
1 前言
新的国家标准《GB/T 5750-2006》和《GB/T 8538-2008》提出31个元素的检测方案,但存在的一些问题需要通过研究予以验证确认,这包括其与《EPA 200.8》的不同或有差异的表述。例如,其一在分析测试中使用的试剂和纯水《GB/T 5750.6-2006》出现明显的差异,《EPA200.8》对试剂做了这样表述:试剂中所含元素杂质将会影响到分析数据的准确性。由于ICP-MS的灵敏度高,要尽可能使用高纯试剂。方法所用酸必须是超高纯级。合适的酸可以通过不同生产厂商购买,也可以采用亚沸点法重蒸馏制备。《EPA200.8》对纯水表述为:本方法所提到的试剂级水是指美国ASTM Ⅰ型水,制备方法:将蒸馏水通过阴离子树脂和阳离子交换树脂混合柱而得。《GB/T 5750.6-2006》认可使用优级纯硝酸,明显不一样。国内优级纯硝酸杂质质量控制不稳定是一方面,其许多微量元素背景浓度超过100μg/L,将直接影响样品结果的可靠性。在纯水方面,不能仅规定电阻率大于18MΩ·cm,而应该说明标准所用纯水来自哪些条件及如何制备。其二,《GB/T 5750.6-2006》没有对氩气提出要求,这是明显漏洞。因为氩气的纯度如果达不到要求,不仅影响仪器运行,还会对分析结果的精密度、准确度产生显著影响。《EPA200.8》对氩气表述为:高纯级(99.99%),如果使用比较频繁,液氩比传统气瓶压缩氩气更经济,且不需要经常更换。《EPA200.8》在另一处这样表述:尤其是,在测定As和Se时会遇到82Kr的干扰,通过使用高纯不含Kr的氩气就能大大降低它的干扰。其三,《GB/T 5750.6-2006》没有说明调谐条件来自哪个型号仪器。因为不同型号的仪器的调谐指标是不一样的。《EPA200.8》则明确其调谐条件来自VG PQ I。其四,在Hg的测定方面,《GB/T 5750.6-2006》仅表述为:“若仪器被污染,应引入含金的溶液清洗。”而且没有说明用多大的浓度清洗。在这方面《EPA200.8》表述的是:“如果采用直接分析步骤测定汞,在内标溶液中加入适量金标准储备液,使最终的空白溶液、校正标准和样品中金浓度达100μg/L。”其五,在内标问题上,《GB/T 5750.6-2006》表述为:“但由于样品中天然存在某些元素而使内标元素的选择受到限制,这些天然存在于样品中的元素将不能做为内标。”《EPA200.8》的主要表述内容与上述有显著不同,如“内标浓度必须足够高,以保证用来校准数据的测定同位素获得好的精密度,如果内标在样品中自然存在,还可使可能的校准偏差降至最低。”再如“同时要扫描样品中被选作内标元素的背景值,防止数据计算时产生偏差。”
在《EPA200.8》中没有明确检测Hg过程不同材料容器对分析结果的影响,由于不同材料的吸附性能不同,对Hg的分析结果产生显著影响已经在实验中得到验证。另外Pb的分析结果也受容器材料的影响。
2 实验原理
电感耦合等离子体质谱(简称 ICP-MS)是等离子体技术与质谱技术相结合分析手段,利用电感耦合等离子体技术做为离子源,以质谱技术做为检测手段,使分析方法具有灵敏度高、分辨率强、检出限低、分析范围宽、分析速度快、检测结果准确的特点。
ICP做为 MS的离子源在于它很好地解决了离子源设计中碰到的两个基本问题:一是获得进样条件和样品激发所需要的可控又无沾污的适当高温环境;二是将样品快速完全地引入到一个对所有期望发生的过程都有足够滞留时间的环境。射频发生器的能量耦合到气流的外环(通常不止使用氩气)形成的环状等离子体可以提供一个气体温度高达 10000 K的区域。在这个区域里能量主要通过热传导传送到冷气流通过的中心通道,气体从石英等离子体炬管以高速沿轴向喷射。仪器系统使用的是直径为 18 mm的炬管,等离子体的频率通常为 27 MHz,入射功率为 1~2 kW。通常使用的通过环状区域的中心通道直径约为 3 mm,气体从炬管口开始几毫秒后到达等离子体,温度由室温升到 8000 K。沿炬管轴向位置发生的过程由气流和电场的场分布所决定。等离子体中的轴向位置由决定电场的场分布感应线圈的外边一圈来定义。最外边的一圈通常离炬管口约 5mm。这个过程在炬管出口处射出原子和离子的混合物及没有分解的残留分子碎片,还有一些没蒸发的粒子的混合物,并伴随大量的氩载气和从环状区域扩散到中心通道的氩气。一旦气体离开火焰,温度骤然下降。当到达采样锥孔位置,通常是离线圈 10~20 mm,气体的温度降至 6000 K或更低。
为了方便进入质谱系统,等离子体炬管在 ICP-AES中是竖着安装的,而在质谱中则水平安装,除此之外和 AES中没什么区别。等离子体火焰围绕着锥口的端部张开,即在锥孔的尖部张开。可以看到中心通道或喷射口气流沿炬管的轴向流动,可以提取很多的气体,环状区的气体沿锥孔的边缘流走。锥孔钻在良导体的锥体的尖部。直径一般使用 1.0~1.2 mm。锥体的寿命是很重要的,其与雾化的酸的类型有极大的关系,如果用 1%硝酸工作的话,全天使用,镍锥体的寿命能使用几个月,但若要使用 10%的硫酸工作的话,可能镍锥体也就仅仅使用几天就需更换。
膨胀到低压区的气体在少于一个锥孔距离内的条件下可以达到超过声速的速度。温度急剧下降 , 可以引起提取气体组成改变的反应即刻停止。随着气压和温度的下降, 动力学能量通过一个称作柱形激波(barrel shock)的冲击波转化成沿轴向的直接流动, 形成有界的自由喷射。第二个冲击波称作马赫盘(Mach disc),是在膨胀的压力比和锥孔直径决定的距离(锥孔后面10 mm处)在轴向形成。因为超出了马赫盘的距离,气流再次变为次声波速度 , 抽进的气体和周围的背景气相混合,在仪器上采用的是把截取锥放在逆向马赫盘的 6.5 mm的距离处。截取锥通常比采样锥的角度更尖一些, 加工成一个尖嘴, 新的截取锥大约“5 µm”宽, 以便使在尖口上形成的冲击波最小。
离开截取锥后, 抽进的气体进入了一个压力足够低的区域, 在这个区域气体平均自由程大于系统的尺寸且气体是随机流动。通常采样锥和截取锥都是在接地电势下工作, 随后形成的离子云的过程完全是一个空气动力学的过程。为了尽可能地把许多离子聚集成一束并通过质量分析器 , 在截取锥之后放置一个静电透镜系统,但通常在透镜系统之前使用一个某种形式的阀门 , 这个阀门能够在分离器之后的通入到高真空区域形成可关闭通道。这个阀门关闭时, 采样锥和截取锥可以在不影响真空压力的情况下取下。如果提取段的压力没降低时这个阀门不能打开。
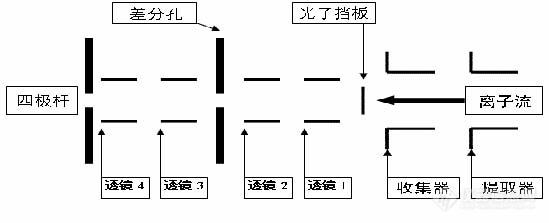
离子透镜系统的形式是根据真空系统的某些使用性能来确定的。质量分析器工作需要的低气压由扩散泵或涡轮分子泵产生,用一个差分抽气小孔隔开,采用两级抽气。离子透镜轴上使用一个光子挡板以阻挡从等离子体来的光子直射到离子检测器形成背景。光子挡板的尺寸由它后面的孔径决定,并在后面孔上投射阴影。
离子透镜的功能是把截取锥后面的离子云尽可能多的在四极质量分析器的入口处形成圆截面的轴向束。四极杆系统,杆的直径为 12~18 mm,长 200 mm。为了维持高质量数的离子的传输,必须使离子以相对慢的速度沿杆的轴迁移,以便离子能有足够的射频场循环数而达到较高的分辨率。这需要离子以较低的离子能量分散进入分析器的杆系统,正常的也就是几个 “eV”。离子在四极杆系统入口处的能量主要是离子源等离子体和(杆)系统之间的直流电势差。因为高于提取孔的等离子体的电势由等离子体的激发和工作参数所决定,所以通常设置一个“调节分析杆” 调节锥口部分的平均直流电压(称为极偏压)。这个极偏压能够使电势落在根据最佳离子能量而设置的等离子体和四极杆之间。极偏压还能用做离子能量粗略测量的阻滞电势。但低能离子可能会由于四极杆末端的弥散场效应受到抑制,这些可以通过使用某种形式的“入口光学系统”使其降至最小。离子首先进入短的入口或四极杆的前端小杆系统。这样就组成了一套和主杆同样直径的一套同轴杆,但仅仅 25 mm长。这些小杆上的 RF电势和主杆相同,而直流成份却被省去了。在主四极杆的出口末端也使用了相似的小四极杆用以改善引出场。
![]()
图 1 离子透镜系统
离子透镜对被提取的离子束几乎没有进行“m/z”的分离。这一过程是通过四极杆质量分析器的质量过滤作用完成的。四极杆质量分析的作用相当一个滤质器,能够通过四极杆质量分析器离子通道的仅仅是一个质量单位的离子。其它质量的离子发生离轴偏转被过滤掉。
![]()
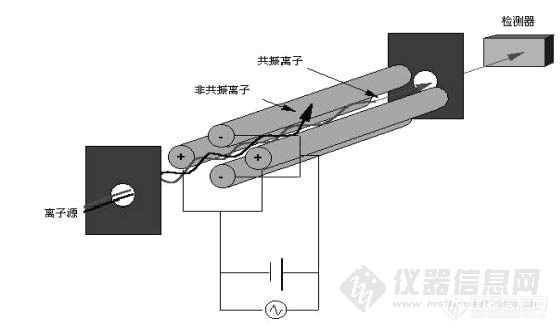
图 2 四极杆原理图
四根笔直的金属或表面镀有金属的极棒与轴线平行并等距悬置。相对的两级连接在一起,幅度为 U和 V的直流和射频电压分别加在每根棒上,一对加正极,一对加负极,每对极棒上所加的电压具有相同的幅度,但 “位差”相差 180度。被分析的离子沿 “轴向”进入四极杆质量分析器的入口,其速度由它们的质量和能量决定。施加的射频电压使所有的离子偏转进入一个振荡路径通过极棒,若适当地选择射频和直流电压,则只有给定的 “m/z”的离子能够得到四极场中的“共振解”而以共振的路径通过极棒,从四极杆质量分析器出口射出,其它的离子将由于无“共振解”而路径过分偏转,与极棒碰撞,并在极棒上被中和掉。
离子在四极杆中的轨迹和离子的传输特性可被相当准确的计算出来。定义,
![]()
式中:U为加在棒上的直流电压,V为加在棒上的射频电压,m/z为离子的质荷比, r为极棒间的内切半径,ω为射频电压频率。以 a为纵坐标,以 q为横坐标,可绘制出离子稳定性图。
![]()
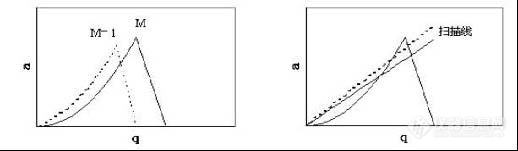
(离子稳定性图) (四极杆扫描原理图)
图 3 四极杆质谱仪扫描图
离子轨迹由 U、V、m/z、r、ω等固定参数决定。大多数 a,q值下,射频和直流电场使离子移出极棒界面,这种轨迹称为不稳定轨迹。只有具有稳定轨迹的离子才能保留在极棒间,这些离子所具有的 a、q值都落在稳定性图的金字塔区域内。
为方便地进行具有不同“m/z”的离子的分离,通常设定 “U/V=常数”,由此可得到一条扫描线,在特定的 U、V值下,扫描线上的每个给定点都对应一个特定的 “m/z”值。因此,改变 U、V值时,a、q值也发生改变,这相当于从一个 “m/z”值沿扫描线移向另一个“m/z”值。
选择扫描线上的 a、q值,相应值 (a、q)处在稳定性图的顶点下,则具有 “m/z=M”的离子将以一个共振的路径通过极棒。在同样条件下的 M-1和 M+1处的邻近离子具有不同的(a、q)值,这些值都在稳定区域外,将不能通过极棒。当有四极杆对不同 m/z值的离子进行分离时,电压 U和 V不断变化,但 U/V保持不变,随着 U、V的不断变化,不同 “m/z”的离子的操作点将移入不同的稳定区域,并通过改变 U、V值获得扫描质谱图。
M与 M-1或 M+1的分离程度取决于扫描线的斜率。如果 U/V增大,扫描线的斜率增大,并靠近稳定区域的顶部,在此情况下通过质量分析器的 “m/z”值的范围变窄,邻近的离子被分离得更完全,仪器的分辨率增大。随着仪器分辨率的增大,稳定区域的面积减少,具有稳定路径的离子数量随之减少,仪器灵敏度急剧下降。
3 实验设备和试剂
3.1 实验设备
- 1.1电感耦合等离子体质谱仪 XSeriesⅡ,赛默飞世尔科技(上海)有限公司;
- MILLIPORE,密理博(上海)贸易有限公司;
- 1.3不间断电源 Prostar SU20K UPS,宝星(佛山)科技发展有限公司;
- PE)塑料样品瓶,规格 100mL;
- 1.5棕色玻璃容量瓶,规格 100mL;
- 1.6聚对苯二甲酸乙二醇酯( PET)塑料瓶,规格 600mL。
- 试剂
- BV-III)500 mL,d(25℃)1.4 g/mL,北京化学试剂研究所;
- BV-III)3.78 L,d(25℃)1.18 g/mL,北京化学试剂研究所;
- 3.2.3纯水( 18.3MΩ·cm),MILLIPORE纯水机;
- 2.4单标标准溶液 ρ(M)=1000µg/mL(金、铑、锂、铍、钒、铬、锰、钴、镍、铜、锌、砷、硒、钼、镉、银、钡、铅、锡、硼、铁),国家钢铁材料测试中心;
- 2.5汞标准溶液 TM-022L,ρ(Hg)=1000µg/mL,北京莱伯泰科科技有限公司(NSI溶液);
- 2.6混合标准溶液 GSBZ50009-88铜铅锌镉镍铬 ρ(M)=0.105~1.49 mg/L,国家环境保护总局标准样品研究所;
- 2.7汞标准溶液 GSB 07-1274-2000,ρ(Hg)=100mg/L,国家环境保护总局标准样品研究所;
- 2.8混合标准溶液 GBW(E)080670,包含元素:K、Na、Ca、Mg、 Fe、Li、Al、V、Cr、Mn、Co、Ni、Cu、Zn、As、Se、Sr、Ag、Cd、Ba、 Bi、Pb、Be、Cs、Rb,ρ(M)=10mg/L,上海市计量测试技术研究院;
- 2.9混合标准溶液 ICPMS-43,包含元素: As、Cd、Cr、Cu、Fe、Pb、 Mn、Ni、Se、V、Zn、Be、Cs、Ce、Mg、Sr、Tl、Dy、Er、Ca、Ag、Al、 B、Ba、K、Na、Eu、Ga、Gd、Ho、La、Lu、Nd、P、S、Pr、Ru、Sm、 Th、Tm、U、Yb,ρ(M)=10µg/mL,北京莱伯泰科科技有限公司( NSI溶液)。
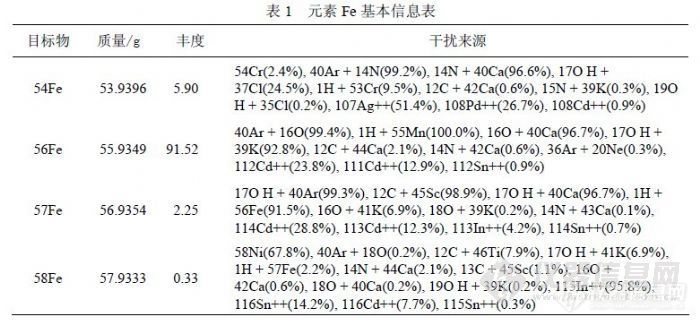
4 检测 Fe的方法研究 4.1 铁的质谱干扰研究《EPA200.8》没有关于 Fe检测的内容,因为其提出时仪器水平处于欧美 1994年水平,《GB/T 5750-2006》提出了 56Fe、57Fe两个同位素,没有说明如何使用这两个同位素及如何消除有关干扰,在本底分子离子干扰方面(ArO+干扰 56Fe)、(ArH+干扰 57Fe)也与《EPA200.8》内容( ArO+干扰 56)、(ArOH+干扰 57)存在区别。![]()
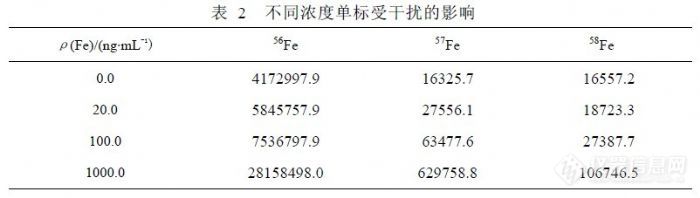
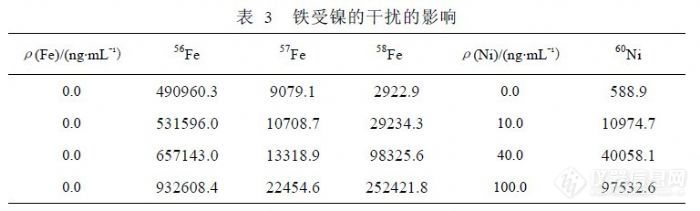
在 ICP-MS分析中,Fe可以检测的同位素有 4个,基本情况如表 1。通常没有干扰的情况下,相同浓度的 Fe同位素测量信号值之间比例关系应与理论丰度关系相同,我们用 1000ng/mL的标准溶液研究 Fe同位素受到其它元素干扰的问题,以确定哪些因素是主要干扰来源。按 Fe同位素丰度关系以 58Fe为基点进行归一化,获得 54Fe、56Fe、57Fe、58Fe的比例关系(17.88:277.33:6.82:1.00),检测 1000ng/mL的 Fe标准溶液获得比例关系 (18.14:263.79:5.90:1.00)。考虑到 Fe标准溶液因纯度问题可能含有少量的其它元素,58Fe的信号值( 106746)与 60Ni(4785)接近, 54Fe(1936520)、 56Fe(28158498)和 57Fe(629759)相对较高,需要扣除 58Ni对 58Fe的干扰,根据 58Ni/60Ni=2.5902的关系,扣除干扰后 58Fe的信号下降 13.14%,同位素的比例关系变为(20.53:298.44:6.68:1.00),再分别扣除 54Cr对 54Fe干扰和 56ArO对 56Fe干扰后比例关系变为( 20.52:254.22:6.68:1.00),从“比例关系”的变化规律可以得出结论,对 54Fe产生干扰的主要因素不是 54Cr,而是 54ArN,其它理论影响因素受产率影响可以不考虑,56Fe和 57Fe在扣除干扰后其比例关系接近理论值说明对 56Fe、57Fe、58Fe的干扰关系定位是合理的,56ArO信号值达到 400万以上,相当于 170ng/mL浓度以上,对低含量 Fe检测影响很大,而 58Fe受到 Ni的严重干扰(因为 58Ni的丰度为67.76%),只有 57Fe受到干扰最小,所以检测 Fe的同位素选择 57Fe是合适的。有文献认为,在 CCT模式下可以消除 56ArO干扰,使 56Fe的检出限达到 0.3pg/mL,但这样的参数对普通矿泉水分析没有实际意义,而模式转换也不适合常规样品检测。![]() 表 2列出了不同浓度的 Fe单标信号值比较,正常情况下 56Fe、57Fe到 58Fe检测灵敏度不同程度下降,受 ArO干扰过大影响,56Fe的线性很差。表 3列出了 60Ni对 58Fe干扰的情况,而 56Fe和 57Fe没有被干扰。
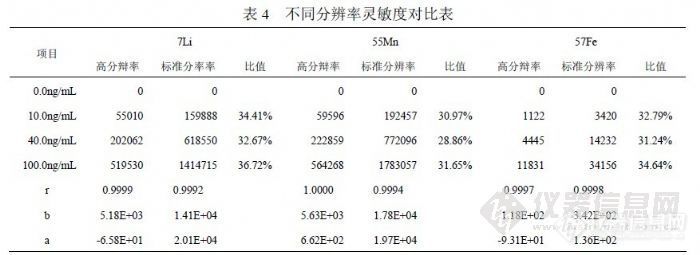
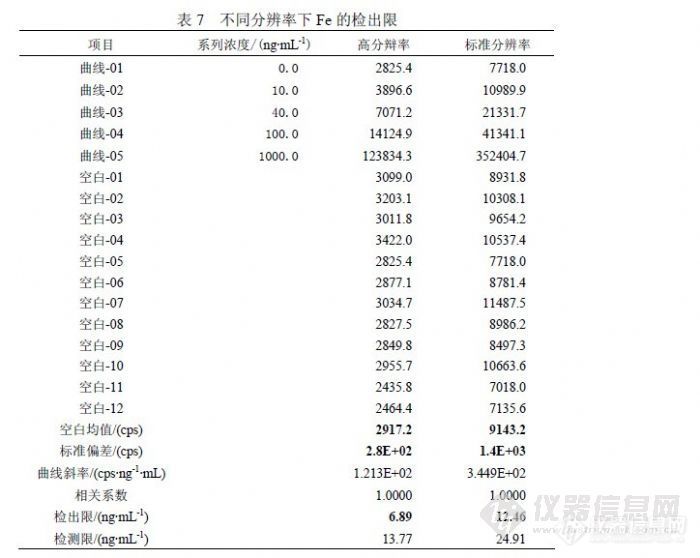
表 2列出了不同浓度的 Fe单标信号值比较,正常情况下 56Fe、57Fe到 58Fe检测灵敏度不同程度下降,受 ArO干扰过大影响,56Fe的线性很差。表 3列出了 60Ni对 58Fe干扰的情况,而 56Fe和 57Fe没有被干扰。![]() 同原子吸收比较,《GB/T 8538-2008》给出 0.3~5.0mg/L检测范围,实际测试往往低于 0.5mg/L时原子吸收就通常报“未检出”,而 ICP-MS在这个浓度范围的灵敏度有非常大的优势。 4.2 铁的分辨率模式研究在 ICP-MS检测过程中,为防止因出现异常情况导致检测器溢出,存在一个“Cross校正”功能,意思是脉冲信号与模拟信号之间关系系数校正,当脉冲检测信号超过设定的临界值(如 350万)时,仪器自动转入模拟状态,使检测器用比较低的模拟电压检测信号,使计数值大幅度下降,达到保护检测器的目的。仪器在每次定量检测前,都会快速预扫描对样品进行估算,决定是否进入模拟状态,但最后显示的信号是模拟信号值与 Cross系数的乘积。所以模拟状态的信号值的准确性取决于 Cross系数是否校正得合理,但无论如何都不及脉冲信号直接检测准确,为使 1.0mg/L以下浓度能够在脉冲状态进行检测,可以使用“高分辩率”模式进行 Fe的分析。按照图 3显示的灵敏度和分辨率关系,采用高分辩率时峰宽由 0.70amu变为 0.35amu,灵敏度下降幅度超过 90%,由于 Fe浓度一般比其它元素高许多,剩余灵敏度仍可以保证 Fe被准确检测。我们利用混合标准溶液在低浓度端统计( 0.0、10.0、40.0、100.0)ng/mL的高分辩率和标准分辨率信号比较,对比标准分辨率信号采用高分辩率时灵敏度下降( 63~72)%,标准曲线线性能够保持,参照 7Li、55Mn变化行为趋于一致,见表 4。
同原子吸收比较,《GB/T 8538-2008》给出 0.3~5.0mg/L检测范围,实际测试往往低于 0.5mg/L时原子吸收就通常报“未检出”,而 ICP-MS在这个浓度范围的灵敏度有非常大的优势。 4.2 铁的分辨率模式研究在 ICP-MS检测过程中,为防止因出现异常情况导致检测器溢出,存在一个“Cross校正”功能,意思是脉冲信号与模拟信号之间关系系数校正,当脉冲检测信号超过设定的临界值(如 350万)时,仪器自动转入模拟状态,使检测器用比较低的模拟电压检测信号,使计数值大幅度下降,达到保护检测器的目的。仪器在每次定量检测前,都会快速预扫描对样品进行估算,决定是否进入模拟状态,但最后显示的信号是模拟信号值与 Cross系数的乘积。所以模拟状态的信号值的准确性取决于 Cross系数是否校正得合理,但无论如何都不及脉冲信号直接检测准确,为使 1.0mg/L以下浓度能够在脉冲状态进行检测,可以使用“高分辩率”模式进行 Fe的分析。按照图 3显示的灵敏度和分辨率关系,采用高分辩率时峰宽由 0.70amu变为 0.35amu,灵敏度下降幅度超过 90%,由于 Fe浓度一般比其它元素高许多,剩余灵敏度仍可以保证 Fe被准确检测。我们利用混合标准溶液在低浓度端统计( 0.0、10.0、40.0、100.0)ng/mL的高分辩率和标准分辨率信号比较,对比标准分辨率信号采用高分辩率时灵敏度下降( 63~72)%,标准曲线线性能够保持,参照 7Li、55Mn变化行为趋于一致,见表 4。![]() 根据上述情况,在 ICP-MS检测 Fe的条件选择应为,在通常情况下选择“高分辩率”模式,若仪器灵敏度降低,可以选择“标准分辨率”检测。 4.3 铁检测的线性范围研究没有具体实验 Fe标准溶液浓度的上限,但实验表明在脉冲计数的范围内 Fe的相关系数>0.999,考虑到 “Main Runs”前进行快速“ Survey Runs”,根据“Survey Runs”的结果确定是进入“Plus”或是“analogue”模式,而快速“Survey Runs”的精密度较差,在计数(3000000~4000000)cps内确定进入哪种模式的不确定性很大,而计数在 3000000cps以下能够明确进入 “Plus”模式,从实验情况确定 Fe标准溶液浓度在“ 2000ng/mL”能够保证信号值低于 3000000cps,所以能够明确的 Fe线性范围在( 0~2000)ng/mL。
根据上述情况,在 ICP-MS检测 Fe的条件选择应为,在通常情况下选择“高分辩率”模式,若仪器灵敏度降低,可以选择“标准分辨率”检测。 4.3 铁检测的线性范围研究没有具体实验 Fe标准溶液浓度的上限,但实验表明在脉冲计数的范围内 Fe的相关系数>0.999,考虑到 “Main Runs”前进行快速“ Survey Runs”,根据“Survey Runs”的结果确定是进入“Plus”或是“analogue”模式,而快速“Survey Runs”的精密度较差,在计数(3000000~4000000)cps内确定进入哪种模式的不确定性很大,而计数在 3000000cps以下能够明确进入 “Plus”模式,从实验情况确定 Fe标准溶液浓度在“ 2000ng/mL”能够保证信号值低于 3000000cps,所以能够明确的 Fe线性范围在( 0~2000)ng/mL。![]()
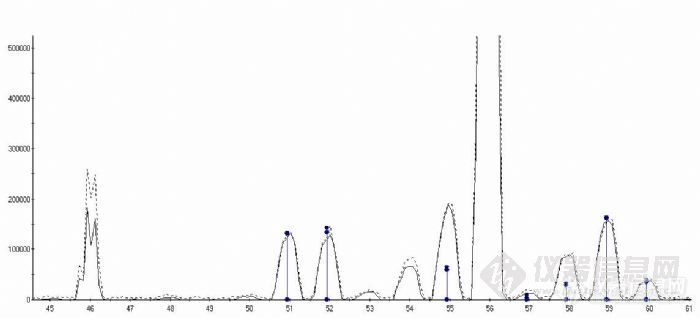
图 4 经 Cross校正后质谱效果图
在“analogue”模式下 Fe的检测线性与“Cross校正”相关,校正结果较好即“analogue质谱峰”与“Plus质谱峰”重合,能够保证线性,否则重合越差线性就越差。图 4中,虚线是“analogue”模式信号,实线是“ Plus”模式信号。在 46、54质量数处校正结果显然不好,其它质量数的校正结果比较好。实际上,同一次校正不同元素的的结果不同。4.4铁的精密度与检出限 4.4.1精密度和准确度实验通过实验统计了不同时间检测的浓度为(10.0、40.0、100.0)ng/mL的标准溶液的相对标准偏差和不确定度。![]()
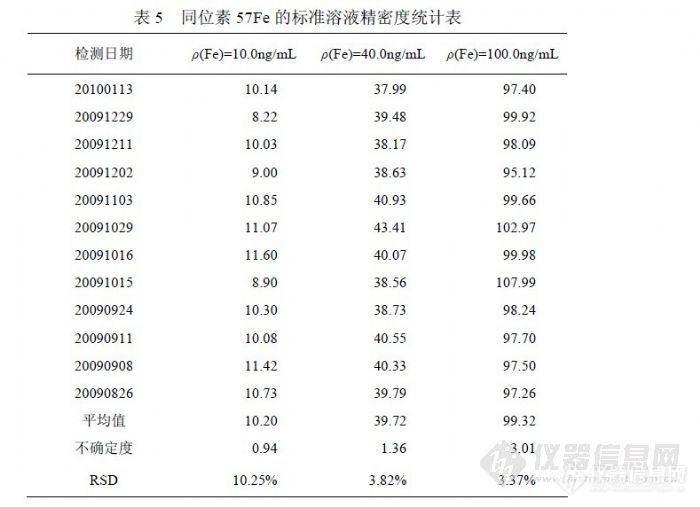
式中:x为平均值,xb为标准值,xi为单次测量值,t(0.99,12) = 3.106。![]() 表5中统计了2009年8月~2010年1月之间的12次标准溶液检测结果,检测主要条件是“高分辩率”模式、质量数 57Fe,标准溶液由 GBW(E) 080670稀释制备,置信水平 0.99。向未知水样中加入 10.0μg的 Fe标准溶液,定容 50.0mL,平行 5份,在检测结果中扣除样品结果后统计加入标准的回收率,见表 6。
表5中统计了2009年8月~2010年1月之间的12次标准溶液检测结果,检测主要条件是“高分辩率”模式、质量数 57Fe,标准溶液由 GBW(E) 080670稀释制备,置信水平 0.99。向未知水样中加入 10.0μg的 Fe标准溶液,定容 50.0mL,平行 5份,在检测结果中扣除样品结果后统计加入标准的回收率,见表 6。 ![]() 4.4.2 检出限实验我们按如下方法完成检出限实验,连续测定十二份空白溶液,计算测得信号的标准偏差,然后通过同时测得的标准曲线计算方法检出限。计算公式如下:
4.4.2 检出限实验我们按如下方法完成检出限实验,连续测定十二份空白溶液,计算测得信号的标准偏差,然后通过同时测得的标准曲线计算方法检出限。计算公式如下: ![]()
式中:DL检出限表示符号,单位“ng·mL”;
r 表示标准曲线的斜率, cps·ng-1·mL;
s 表示测量信号的标准偏差, cps。
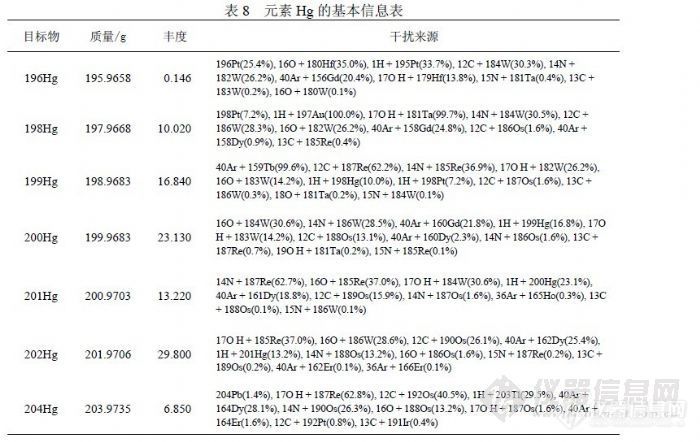
通过实验,比较了“高分辩率”模式和“标准分辨率”模式的检出限参数,在“高分辩率”模式下能够获得更好的检出限指标。可能的原因是,由于峰宽变窄后,虽然灵敏度下降,但质谱峰依然符合高斯分布,因此信号离散度较小,而检出限高低主要取决于空白信号的离散度(样本标准偏差)。![]() 5 检测 Hg的方法研究在传统检测 Hg的方法主要有原子吸收法和原子荧光法,国外多使用氢化物发生原子吸收法,国内多采用氢化物发生原子荧光法,当前国家标准也主要推荐原子荧光法检测 Hg。在水分析中, Hg的含量一般很低,非常容易受检测环境和检测条件等因素影响。例如原子荧光法检测 Hg的过程中需要加入硼氢化钾、重铬酸钾、硝酸、盐酸和不同纯度的纯水等,它们合起来将产生试剂空白,按现行检测方法,无论扣与不扣都会使分析结果发生偏离,而较大背景空白因修正标准偏差使检出限向偏低方向偏离,从而产生错觉。进行 Hg的分析方法研究也是力图在 ICP-MS仪器检测过程中消除这些能够影响分析结果的因素。 5.1 汞的同位素(质量数)选择关于 Hg的基本信息见表 8,可供 ICP-MS检测的同位素计有 196Hg、 198Hg、199Hg、200Hg、201Hg、202Hg、204Hg七个,但通常见于文献的仅有 200Hg、201Hg、202Hg三个,可能原因是 Hg在水中浓度很低,信号值一般很小,而 196Hg、198Hg、204Hg的丰度较小,相对灵敏度也就很低,检测低浓度 Hg比较困难;对于 199Hg其存在“ 40Ar+159Tb”的干扰,一但样品中 Tb含量较高就会对 Hg形成严重干扰,另外 204Hg容易受到 204Pb的干扰,相比之下 200Hg、201Hg、202Hg基本不受其它元素干扰,并且有较高丰度。
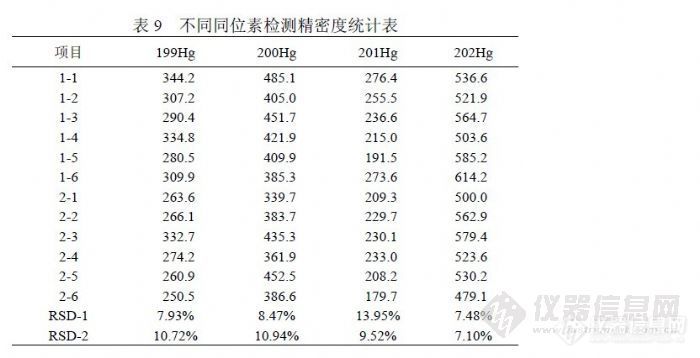
5 检测 Hg的方法研究在传统检测 Hg的方法主要有原子吸收法和原子荧光法,国外多使用氢化物发生原子吸收法,国内多采用氢化物发生原子荧光法,当前国家标准也主要推荐原子荧光法检测 Hg。在水分析中, Hg的含量一般很低,非常容易受检测环境和检测条件等因素影响。例如原子荧光法检测 Hg的过程中需要加入硼氢化钾、重铬酸钾、硝酸、盐酸和不同纯度的纯水等,它们合起来将产生试剂空白,按现行检测方法,无论扣与不扣都会使分析结果发生偏离,而较大背景空白因修正标准偏差使检出限向偏低方向偏离,从而产生错觉。进行 Hg的分析方法研究也是力图在 ICP-MS仪器检测过程中消除这些能够影响分析结果的因素。 5.1 汞的同位素(质量数)选择关于 Hg的基本信息见表 8,可供 ICP-MS检测的同位素计有 196Hg、 198Hg、199Hg、200Hg、201Hg、202Hg、204Hg七个,但通常见于文献的仅有 200Hg、201Hg、202Hg三个,可能原因是 Hg在水中浓度很低,信号值一般很小,而 196Hg、198Hg、204Hg的丰度较小,相对灵敏度也就很低,检测低浓度 Hg比较困难;对于 199Hg其存在“ 40Ar+159Tb”的干扰,一但样品中 Tb含量较高就会对 Hg形成严重干扰,另外 204Hg容易受到 204Pb的干扰,相比之下 200Hg、201Hg、202Hg基本不受其它元素干扰,并且有较高丰度。![]() 我们在样品分析中观察了几个主要同位素的精密度情况,由于精密度与灵敏度相关,出现因灵敏度下降而导致精密度下降的趋势,相比之下 202Hg更稳定。样品分 2次进样每次进样连续检测 6次,分别统计 RSD,统计结果见表 9。
我们在样品分析中观察了几个主要同位素的精密度情况,由于精密度与灵敏度相关,出现因灵敏度下降而导致精密度下降的趋势,相比之下 202Hg更稳定。样品分 2次进样每次进样连续检测 6次,分别统计 RSD,统计结果见表 9。![]() 5.2 汞的记忆效应研究 5.2.1 内标溶液加入金对清洗效果的影响《GB/T 8538-2008》(《GB/T 5750.6-2006》)提出:“若仪器被污染,应引入含金的溶液清洗。”但没有说明用多大的浓度清洗。在这方面《EPA200.8》表述为:“如果采用直接分析步骤测定汞,在内标溶液中加入适量金标准储备液,使最终的空白溶液、校正标准和样品中金浓度达 100μg/L。”
5.2 汞的记忆效应研究 5.2.1 内标溶液加入金对清洗效果的影响《GB/T 8538-2008》(《GB/T 5750.6-2006》)提出:“若仪器被污染,应引入含金的溶液清洗。”但没有说明用多大的浓度清洗。在这方面《EPA200.8》表述为:“如果采用直接分析步骤测定汞,在内标溶液中加入适量金标准储备液,使最终的空白溶液、校正标准和样品中金浓度达 100μg/L。” ![]()
图 5 内标溶液加入 Au对清洗效果的影响
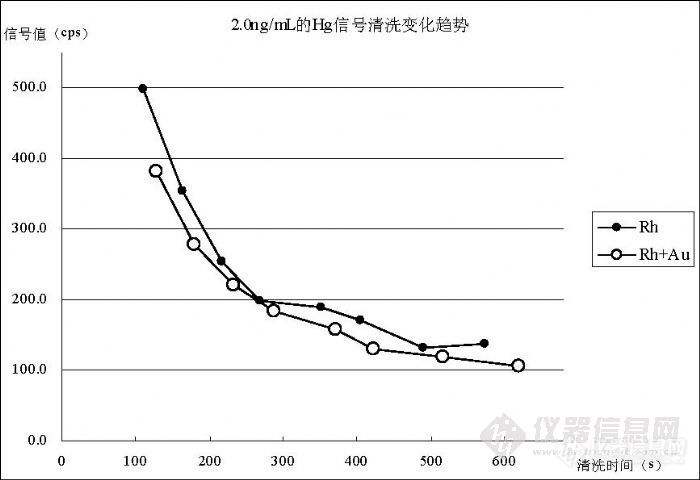
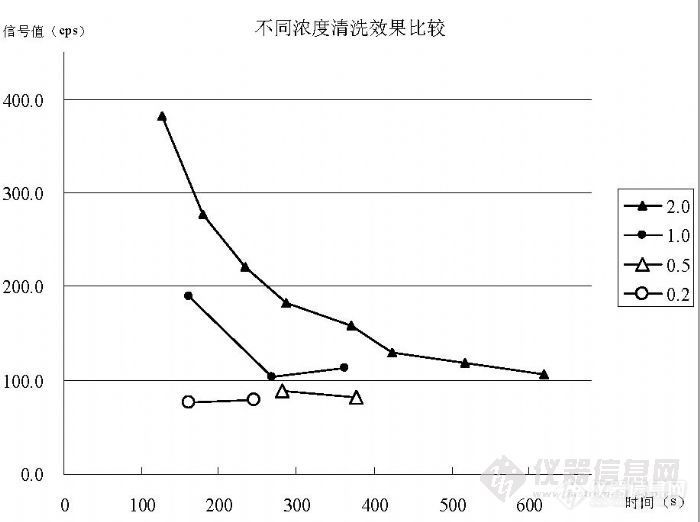
我们采用在内标溶液中加入“200ng/mL的 Au标准溶液”的方法,然后以“在线内标”的方式进行检测,通过与没有加入 Au标准溶液的内标溶液进行比较,获得图 5所示效果。在 Rh内标溶液条件下和 Rh+Au内标溶液条件下分别检测浓度“2.0ng/mL的 Hg标准溶液”后,分别获得“7382.1cps”和“7380.1cps”的信号值,用同一的 2%硝酸溶液进行清洗,清洗过程为 4次进样采集 40个数据,图中每个数据点为 5个数据的平均值。最终, Rh条件下在 490s时达到 131.6cps,在 574s时达到 137.3cps;在 Rh+Au条件下 423s时达到 129.1cps,在 517s时达到 118.6cps,在 619s时达到 106.1cps。数据说明,Rh+Au内标溶液的清洗效果比 Rh的效果好。 5.2.2 不同浓度标准溶液的清洗效果比较选择(0.20、0.50、1.0、2.0)ng/mL的标准溶液进行检测后清洗,图 6说明了清洗效果的趋势。![]()
图 6 不同浓度标准溶液的清洗效果
浓度为 2.0ng/mL的标准溶液经 619s达到 106.1cps,最低 88.1cps达到空白溶液的背景水平;浓度为 1.0ng/mL的标准溶液经 363s达到 113.4cps,最低96.2cps接近空白溶液的背景值水平;浓度为0.50ng/mL的标准溶液经281s达到 88.4cps,经 377s稳定在 82.5cps空白溶液背景值水平,最低能够达到 - 6cps;浓度为 0.20ng/mL的标准溶液经 161s达到 76.8cps,经 246s稳定在
- 3cps水平,最低 59.9cps,处于空白溶液背景值水平。比较的数据说明浓度在 0.5ng/mL以上需要较长时间(>5min)清洗才能接近空白溶液背景水平,不对后面样品产生干扰;浓度在 0.2ng/mL以下只要清洗时间超过 3min就不会对后面的样品产生影响。
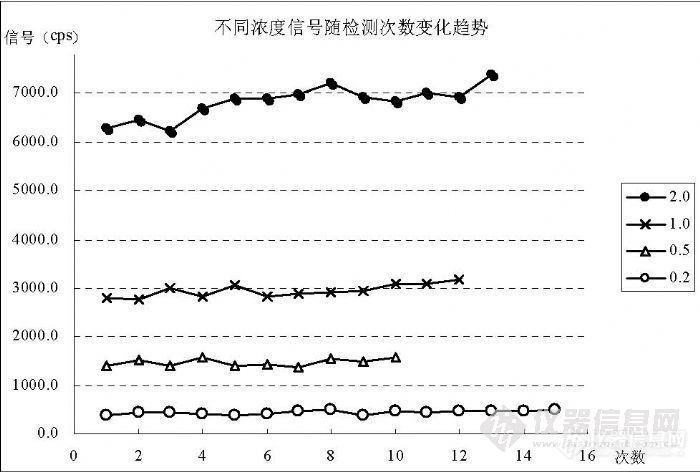
5.2.3 不同浓度标准溶液的记忆效应在实验中观察到,Hg的标准溶液不像其它元素那样,随进样时间延长而趋于稳定,而是随进样时间增加其计数信号呈上升趋势,不同浓度其程度不一样。图 7的结果说明了这一点。![]()
图 7 Hg标准溶液记忆效应变化趋势
图中显示,浓度 0.20ng/mL的标准溶液记忆效应并不明显,而浓度0.50ng/mL的标准溶液已经显示出随连续进样次数增加信号值上升趋势;浓度 1.0ng/mL和 2.0ng/mL的记忆效应显著增强,只是 1.0ng/mL的程度没有2.0ng/mL的大。由此说明在Hg的检测过程中,采集数据的次数和时间不宜过长,应控制在有限次(一般最多 5次,时间 150s)之内,更多次数的采集应在中间插入空白溶液清洗。在 0.20ng/mL以下的低浓度记忆效应趋势不显著,可以直接进行多次采集。Hg的记忆效应与浓度相关。 5.3 容器材料对检测结果的影响在实验中观察到,采用不同的盛装容器将对分析结果产生显著影响。塑料容器可能由于吸附作用使 Hg的信号被抑制,高浓度受到抑制的程度比低浓度的小,高密度塑料材料抑制的程度比低密度材料的小,低密度塑料材料容器的抑制程度相互间不一致,最好使用玻璃容器盛装测试溶液。表 10的数据说明这个趋势。在上面表格中,统计了浓度为(0.00、0.10、0.20、0.50)ng/mL的 Hg标准溶液配制在不同容器中进行检测的信号值,均为当天配制并检测的结果,表中数据均为相同检测条件。由于原使用塑料瓶配制标准曲线,但多次配制的结果出现较大差异,并且无法获得稳定的线性,所以制定了这个条件实验,最终查出问题原因。使用玻璃容量瓶配制标准曲线,能够获得稳定的检测结果,线性也符合检测要求。![]() 5.4 汞的灵敏度与其它元素的关系通过实验观察了在检测过程中,因仪器波动对检测信号产生影响时 Hg与内标 Rh之间的变化趋势。在图 8中统计了 33个连续的检测点,1~5是空白溶液进样后连测 5次,6~10是 0.10ng/mL的 Hg标准溶液进样后连测 5次,11~15是0.20ng/mL的Hg标准溶液进样后连测5次,16~20是0.50ng/mL的 Hg标准溶液进样后连测 5次,21~28是空白溶液进样后连测 8次,29~ 33是 0.50ng/mL的 Hg标准溶液进样后连测 5次,分别以每次进样第一个检测点为基准点做归一化处理,比较同一检测点各比值变化趋势。
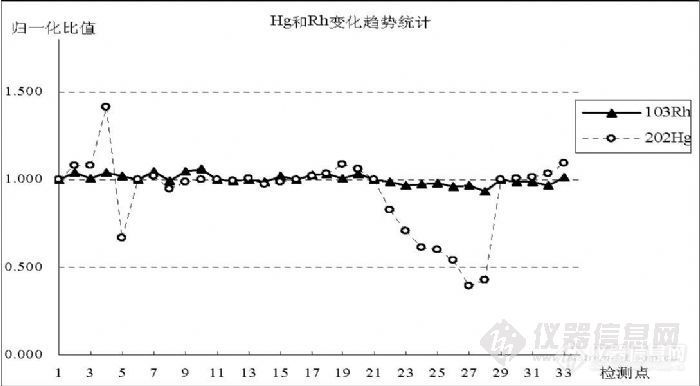
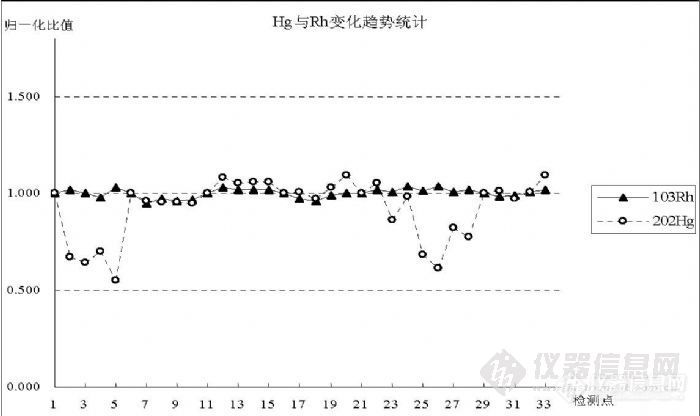
5.4 汞的灵敏度与其它元素的关系通过实验观察了在检测过程中,因仪器波动对检测信号产生影响时 Hg与内标 Rh之间的变化趋势。在图 8中统计了 33个连续的检测点,1~5是空白溶液进样后连测 5次,6~10是 0.10ng/mL的 Hg标准溶液进样后连测 5次,11~15是0.20ng/mL的Hg标准溶液进样后连测5次,16~20是0.50ng/mL的 Hg标准溶液进样后连测 5次,21~28是空白溶液进样后连测 8次,29~ 33是 0.50ng/mL的 Hg标准溶液进样后连测 5次,分别以每次进样第一个检测点为基准点做归一化处理,比较同一检测点各比值变化趋势。![]()
图 8 Hg信号与内标 Rh变化趋势比较图之一
统计结果表明,受前面高浓度信号影响,空白溶液处于清洗过程中,信号持续下降,Hg与 Rh变化趋势不符,但排除这一影响因空白信号值较小离散度大而与 Rh产生偏离,图 9是相同实验过程但没有受到高浓度信号影响的结果;浓度 0.10ng/mL和 0.20ng/mL的变化趋势与 Rh相同,说明内标能够有效校正仪器波动对检测信号产生的影响;浓度 0.50ng/mL的两段重复结果说明 Hg信号变化趋势与 Rh相符,但受记忆效应影响呈现逐渐升高趋势,每段最后两个检测点表现得比较明显;中间 21~28是 8个空白清洗结果,呈下降趋势。图 9是和图 8在不同时间进行的相同过程实验,使用了另外一份溶液完成,表现了相近的变化趋势,都说明 Rh能够有效校正 Hg在检测过程中的信号波动。 ![]()
图 9 Hg信号与内标 Rh变化趋势比较图之二
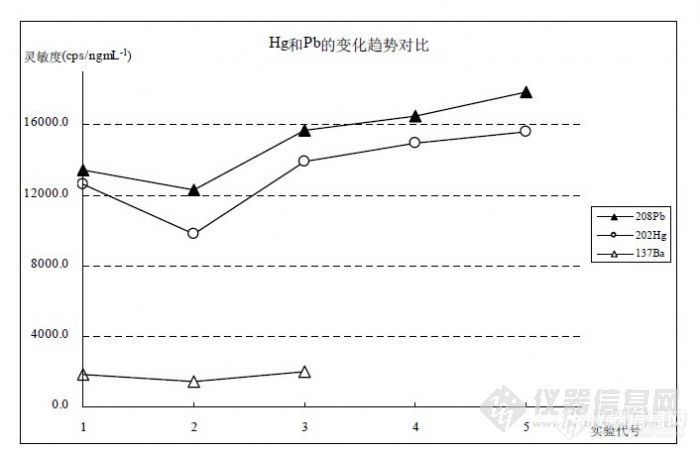
在图 10的对比曲线中,从 1~5的实验代号分别表示 2009年 12月 29日、12月 2日、11月 3日、10月 30日、10月 29日的实验,每个实验统计了灵敏度(cps·ng-1·mL)进行比较,可以观察到 Hg、Pb、Ba都有相同的变化趋势,由于 202Hg与 208Pb质量数接近,在灵敏度高低和变化幅度方面两者的程度较为接近。综合 10月 29日到 12月 29实验灵敏度数据,经对 Pb归一化处理后,统计 202Hg对 208Pb比值为(0.8469±0.2422),同时 137Ba对 208Pb比值为(0.1308±0.0079)。
![]()
图 10 灵敏度 Hg 与 Pb、Ba 变化趋势对比统计
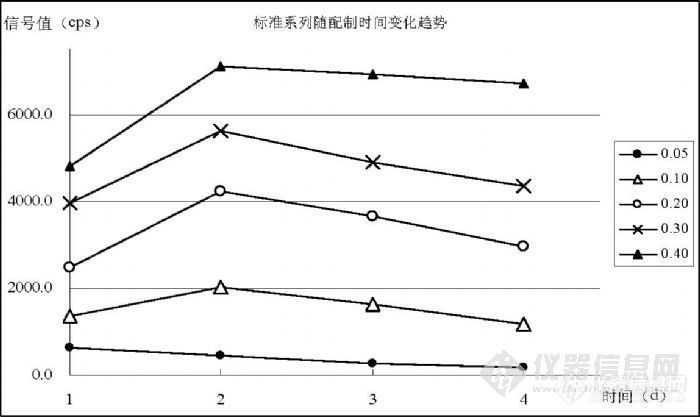
5.5 汞标准溶液保存条件的研究通常预测标准溶液应随存放时间的延长浓度呈下降趋势,Hg的稳定性较差,原预计同一样品随存放时间延长检测灵敏度应下降,但实验中发现的现象并非如此。图 11是配制浓度(0.05、0.10、0.20、0.30、0.40)ng/mL的 Hg标准溶液在不同放置时间中跟踪检测所表现出的信号变化趋势,其中“ 1”表示溶液配制当天,“2”是第二天,依此类推;由检测时间不同产生的灵敏度差异通过内标进行校正,并以 137Ba、208Pb进行监控,能够保证进行比较的数据之间灵敏度水平相当。![]()
图 11标准溶液随配制时间延长变化趋势
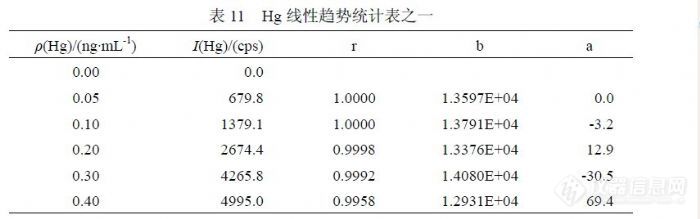
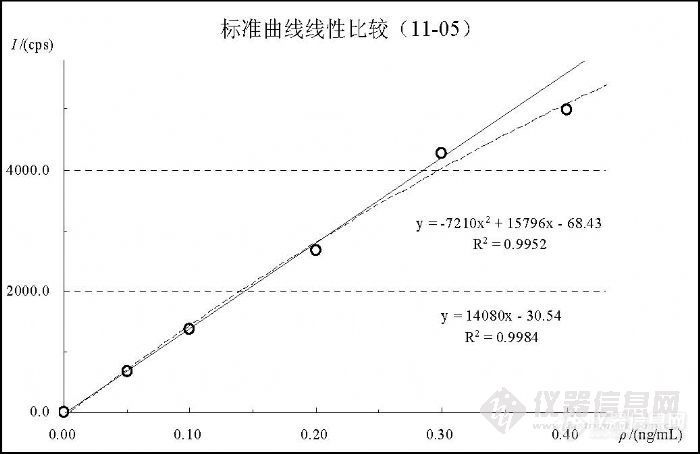
图中各浓度变化趋势表明,Hg的标准曲线必须使用玻璃容量瓶现用现配,放置超过 1天各浓度点将发生不同程度和不同趋势的变化。至于发生这种变化的原因目前还没有确定。 5.6 汞线性范围研究对于 Hg检测的线性范围,EPA200.8认为在低于 5ng/mL的范围内都是一致的,GB/T5750.6-2006提出了( 0.0、0.1、0.5、1.0、1.5、2.0)ng/mL标准系列配制浓度,但在实验中观察到不一样的情况。在表 11和图 12是 11月 5日实验统计的标准曲线变化,表 12、表 13和表 14是 11月 16日实验统计的标准曲线变化情况。在 11月 5日实验观察到,浓度在( 0.00~0.30)ng/mL时能够保持较好的线性和截距(如表 11),当计入 0.40ng/mL时相关系数出现明显的变差并且截距出现较大上升,基于曲线原点的上述幅度相当于一个方法检出限水平,足以对低浓度检测结果产生影响。在图 12中观察到符合 0.40ng/mL变化规律的二次拟合曲线与 0.30ng/mL的一次回归曲线的比较情况,在浓度点0.20ng/mL之前两条曲线符合较好,约在 0.25ng/mL以后出现比较明显分离,在 0.40ng/mL处出现显著的偏低趋势。![]()
![]()
图 12 Hg标准曲线线性影响效果图
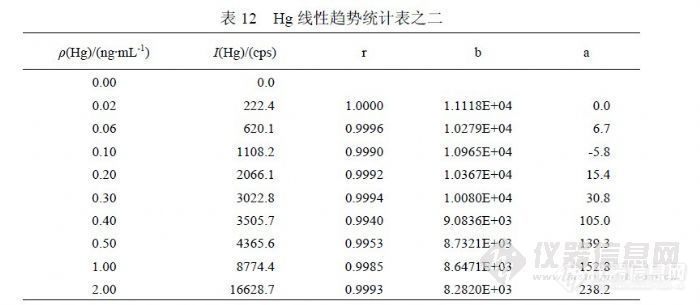
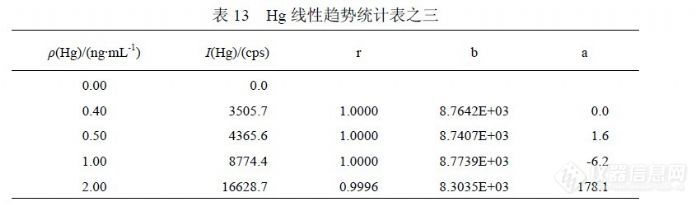
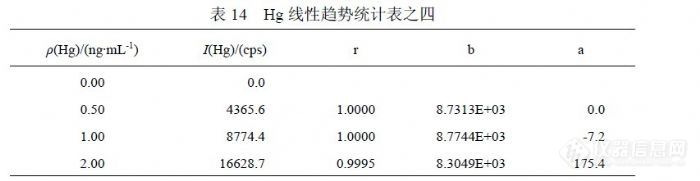
在 11月 16日实验中增加了浓度点,表 12统计的是( 0.00~2.0)ng/mL标准曲线的相关系数、斜率、截距,各参数是对应浓度点到 0.00ng/mL范围的统计结果。可以观察到,自 0.40ng/mL以后曲线参数向“变差”方向发生偏离,呈增强趋势,其截距升高幅度超过检出限信号值水平,足以对低浓度检测结果产生显著影响。表 13是扣除( 0.02~0.30)ng/mL浓度点后统计的曲线参数,其在另一个灵敏度下呈现较好线性,比较灵敏度之间关系,将有可能使同一样品的检测结果升高 20%。注意到 2.0ng/mL对曲线产生较差影响,其对截距产生的作用使其适合检测 0.20ng/mL以上浓度的样品。表 14是在表 13的基础上扣除 0.40ng/mL浓度点后统计曲线参数,结果与表 13差异不明显。![]()
![]()
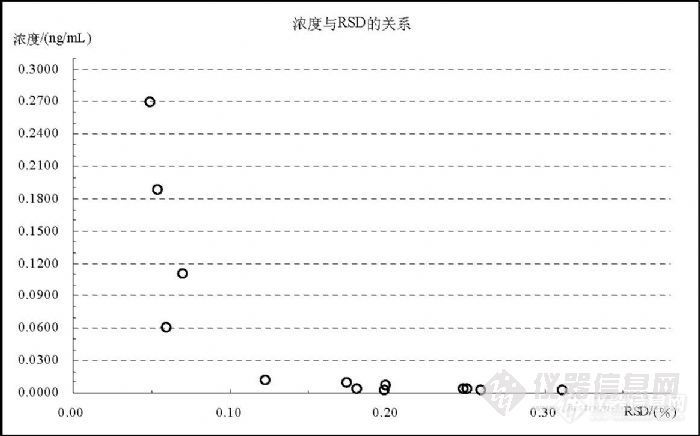
![]() 上述研究表明,在直接检测水中 Hg的过程中,检测范围可以在( 0~2) ng/mL,当浓度低于“ 0.25 ng/mL”时应采用( 0.00、0.05、0.10、0.20、0.30) ng/mL的标准曲线,当浓度高于(含) “0.25 ng/mL”时应采用( 0.00、0.40、0.50、1.0、2.0)ng/mL的标准曲线检测。 5.7 汞的测量精密度和准确度研究通过标准溶液和样品检测的实验数据统计了解到,Hg的检测精密度与其浓度相关,浓度越低精密度越差,浓度越高精密度越好,图 13表明了这个趋势。图中离 10%较近的点浓度 0.0115ng/mL,其 RSD为 12.31%,在实验中 RSD超过 10%的浓度均低于 0.0200 ng/mL,图中趋势在 RSD为 10%的临界浓度约在 0.03 ng/mL左右。
上述研究表明,在直接检测水中 Hg的过程中,检测范围可以在( 0~2) ng/mL,当浓度低于“ 0.25 ng/mL”时应采用( 0.00、0.05、0.10、0.20、0.30) ng/mL的标准曲线,当浓度高于(含) “0.25 ng/mL”时应采用( 0.00、0.40、0.50、1.0、2.0)ng/mL的标准曲线检测。 5.7 汞的测量精密度和准确度研究通过标准溶液和样品检测的实验数据统计了解到,Hg的检测精密度与其浓度相关,浓度越低精密度越差,浓度越高精密度越好,图 13表明了这个趋势。图中离 10%较近的点浓度 0.0115ng/mL,其 RSD为 12.31%,在实验中 RSD超过 10%的浓度均低于 0.0200 ng/mL,图中趋势在 RSD为 10%的临界浓度约在 0.03 ng/mL左右。![]()
图 13 Hg不同检测浓度的 RSD趋势图
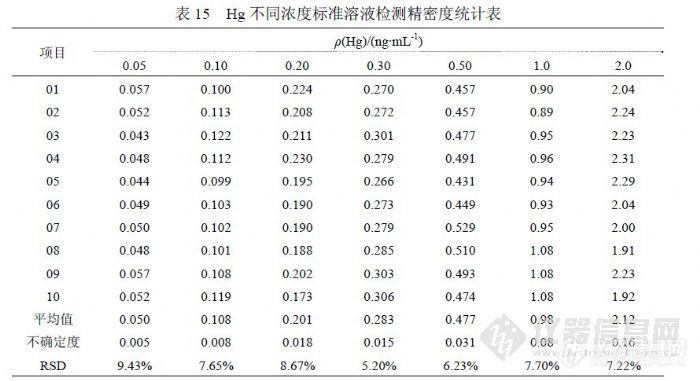
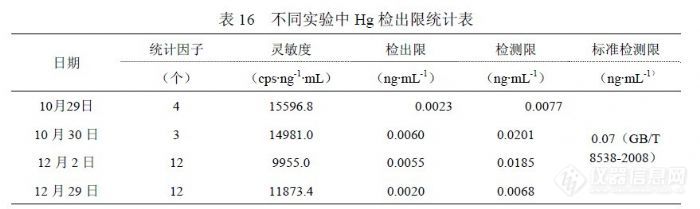
对 2009年 11~12月的部分样品进行统计,样品浓度范围在(0.0028~ 0.0244)ng/mL,RSD在(10.74~66.02)%。我们对部分实际检测的样品进行统计,浓度超过检测限(约 0.02 ng/mL)应能够满足 RSD在 30%以内要求,但 20%不能保证,对于某些不管浓度高低均报出检测结果的情况,当浓度在检测限(约 0.02ng/mL)以下时, RSD超过 30%难以避免(浓度在 0.010 ng/mL~0.020 ng/mL的 RSD为 11.04%~21.19%,浓度在 0.010 ng/mL以下的 RSD为 10.74%~66.02%)。问题比较棘手的是准确度控制方面,因为目前国内没有可用于 ICP-MS检测水中 Hg的标准物质(能够直接上机检测的溶液),无法使用标准物质对 Hg的检测过程进行监控;浓度最低的标准物质是模拟天然水中汞标准物质 GBW(E)080392,浓度( 0.010±5%)mg/L,介质 3%HNO3+0.05%K2Cr2O7,其浓度比《 GB/T 8538-2008》限定 Hg标准曲线最高 2.0 ng/mL高 5倍,比实际使用标准曲线浓度点高 50倍,比多数样品的分析结果能高出 500倍以上,实际上对样品分析的准确度控制不可靠;标准溶液经逐级稀释达到接近样品浓度,由于加入实验室空白并接受实验室环境影响,实际影响检测准确度仍取决于实验室具体检测条件;所以在 Hg的检测中关于插入标准物质及标准物质证书的要求没有意义。在加标回收方面,浓度在 1000 ng/mL以下的回收率允许限(80~120)%,实际相当于检测精密度(RSD)控制在 20%以内,如此一来就需要加标量不能低于 0.10ng/mL,这个浓度高于检测限 5倍,使准确度的控制失去意义,因为检测限的数量级正好与 0.10ng/mL的不确定度水平相当。一般有证 Hg标准物质的浓度为“100μg/mL”或“1000μg/mL”,以最简捷的 100倍稀释方法达到“0.10 ng/mL”需要进行 3个过程,每个过程使用的 A级量具(容量瓶和移液管)控制体积误差是“±1%”,累计误差“±6%”,再加上标准溶液自带“±5%”的相对偏差合计“±11%”,在实验中统计的“0.10 ng/mL”检测 RSD就是“±10%”,所以全程合计 RSD就是 “±21%”,如果加标后合计浓度低于“0.10 ng/mL”水平则统计回收率超过允许限的可能性就非常高。考虑样品实际浓度,加标量在“(0.02~0.05) ng/mL”比较合适,这个浓度的 RSD为 30%,考虑稀释过程的相对偏差合计“±13%”,Hg微量检测的加标回收率允许限在(55~145)%更为合理。![]() 表 15是统计 10月 29日、10月 30日、11月 3日、12月 2日、12月 29日实验中各浓度点合计检测结果,统计方法按(4.2.1),其中 t(0.99,10)=3.250,置信水平 99%,测量次数 10次。 5.8汞的检出限和加标回收率 5.8.1汞的方法检出限和检测限在多个实验中统计了检出限参数,目的在于确切了解方法检出限的实际水平,从实验中了解到检出限主要与灵敏度水平和统计检出限的空白溶液制备水平有关,空白溶液制备过程中所使用的水、硝酸纯度越高和受环境影响越小则越容易获得较好的检出限,因为这样能够获得较低的信号值和更好的精密度。在表 16中,统计因子说明了参与检出限计算的空白溶液数量,检出限按(4.4.2)方法采用 3倍标准偏差统计,由于 Hg一般检测浓度较低,为加强检测结果的可靠性,检测限没有按通常 6倍标准偏差统计,而是采用 10倍标准偏差进行统计。
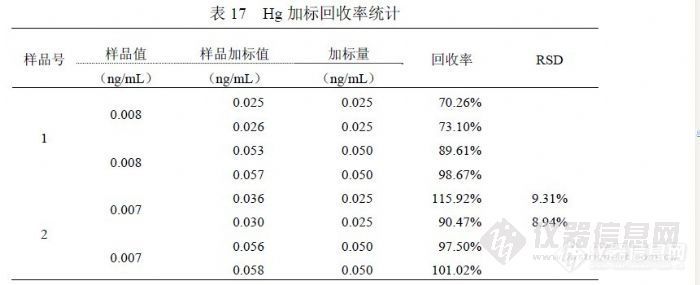
表 15是统计 10月 29日、10月 30日、11月 3日、12月 2日、12月 29日实验中各浓度点合计检测结果,统计方法按(4.2.1),其中 t(0.99,10)=3.250,置信水平 99%,测量次数 10次。 5.8汞的检出限和加标回收率 5.8.1汞的方法检出限和检测限在多个实验中统计了检出限参数,目的在于确切了解方法检出限的实际水平,从实验中了解到检出限主要与灵敏度水平和统计检出限的空白溶液制备水平有关,空白溶液制备过程中所使用的水、硝酸纯度越高和受环境影响越小则越容易获得较好的检出限,因为这样能够获得较低的信号值和更好的精密度。在表 16中,统计因子说明了参与检出限计算的空白溶液数量,检出限按(4.4.2)方法采用 3倍标准偏差统计,由于 Hg一般检测浓度较低,为加强检测结果的可靠性,检测限没有按通常 6倍标准偏差统计,而是采用 10倍标准偏差进行统计。![]() 多次统计表明,ICP-MS技术检测饮用水中 Hg的检出限在(0.002~ 0.007)ng/mL范围内,优于《 GB/T 8538-2008》提出的 0.07ng/mL水平,确定能够提供可靠结果的界限参数检测限水平在(0.007~0.03)ng/mL级别。 5.8.1 汞的加标回收率同位素 202Hg的加标回收率实验按如下方法进行,选择两个未知样品分别作平行双份检测样品结果,另取样品加入 Hg标准溶液,每个样品都做 0.025ng/mL和 0.050ng/mL的标准加入,每个浓度的加入都做平行双份,上述合计样品 12份;使用 10.0ng/mL的 Hg标准溶液,样品定容体积 100.0mL,不加硝酸保持样品原来酸度。对 2号样品 0.025ng/mL加标量的双份样品各进行“1次进样连续采集 6次”的过程 1次统计其检测精密度。统计结果见表 17。
多次统计表明,ICP-MS技术检测饮用水中 Hg的检出限在(0.002~ 0.007)ng/mL范围内,优于《 GB/T 8538-2008》提出的 0.07ng/mL水平,确定能够提供可靠结果的界限参数检测限水平在(0.007~0.03)ng/mL级别。 5.8.1 汞的加标回收率同位素 202Hg的加标回收率实验按如下方法进行,选择两个未知样品分别作平行双份检测样品结果,另取样品加入 Hg标准溶液,每个样品都做 0.025ng/mL和 0.050ng/mL的标准加入,每个浓度的加入都做平行双份,上述合计样品 12份;使用 10.0ng/mL的 Hg标准溶液,样品定容体积 100.0mL,不加硝酸保持样品原来酸度。对 2号样品 0.025ng/mL加标量的双份样品各进行“1次进样连续采集 6次”的过程 1次统计其检测精密度。统计结果见表 17。![]() 表中 RSD统计说明了仪器在相应浓度(0.030ng/mL左右)所具有的精密度水平,属于最基础的参数。浓度为 0.025ng/mL的回收率结果在( 70.26~ 115.92)%范围,浓度 0.050ng/mL的回收率结果在( 89.61~101.02)%范围。由于浓度较低,相应精密度较差,统计的回收率结果没有能够控制在( 80~ 120)%范围内。 6 检测多元素的方法研究使用 ICP-MS技术检测饮用水中微量元素在国内外被普遍采用,《EPA200.8》提出了 21个元素(有关检出限参数均采用 μg/L以下同)(27Al 0.02、23Sb 0.008、75As 0.02、137Ba 0.03、9Be 0.02、111Cd 0.02、52Cr 0.04、59Co 0.002、63Cu 0.004、206+207+208Pb 0.015、55Mn 0.007、202Hg 0.2、98Mo 0.005、 60Ni 0.07、82Se 1.3、107Ag 0.004、205Tl 0.014、232Th 0.005、238U 0.005、51V 0.006、 66Zn 0.07),《GB/T 8538-2008饮用天然矿泉水检验方法》(暨《GB/T 5750-2006》)提出 31个元素( 107Ag 0.03、27Al 0.6、75As 0.09、11B 0.9、135Ba 0.3、9Be 0.03、40Ca 6.0、111Cd 0.06、59Co 0.03、52Cr 0.09、63Cu 0.09、56Fe 0.9、 39K 3.0、7Li 0.3、24Mg 0.4, 55Mn 0.06、98Mo 0.06、23Na 7.0、60Ni 0.07、208Pb 0.07、121Sb 0.07、77Se 0.09、88Sr 0.09、118Sn 0.09、232Th 0.06、203Tl 0.01, 48Ti 0.04、235U 0.04、51V0.07、66Zn 0.8、202Hg0.07),我们利用 PQ ExCell ICP-MS自二○○二年以来进行了检测饮用水中微量元素方法研究,提出了 15个元素(7Li 0.051、27Al 1.8、51V 0.044、52Cr 0.23、55Mn 0.20、59Co 0.17、60Ni 0.99、 65Cu 0.17、66Zn 0.42、82Sr 0.052、98Mo 0.037、107Ag 0.013、114Cd 0.022、137Ba 0.17、208Pb 0.088),现在仪器更换为 XSeries2后在性能方面发生很多变化,根据分析测试需要在检测项目方面也发生了很多变化,需要通过方法研究解决这些变化,另外在《 EPA200.8》和《GB/T 8538-2008》之间也存在诸多不同之处,需要通过研究进行确认。在国内已有很多 ICP-MS检测水中微量元素的文献发表,检测元素最多的已经达到 44项,检测方法存在很多不同的地方,对于这些文献提出的结论,是否值得我们借鉴,需要通过研究和实验进行验证。 6.1多元素的检测条件研究
表中 RSD统计说明了仪器在相应浓度(0.030ng/mL左右)所具有的精密度水平,属于最基础的参数。浓度为 0.025ng/mL的回收率结果在( 70.26~ 115.92)%范围,浓度 0.050ng/mL的回收率结果在( 89.61~101.02)%范围。由于浓度较低,相应精密度较差,统计的回收率结果没有能够控制在( 80~ 120)%范围内。 6 检测多元素的方法研究使用 ICP-MS技术检测饮用水中微量元素在国内外被普遍采用,《EPA200.8》提出了 21个元素(有关检出限参数均采用 μg/L以下同)(27Al 0.02、23Sb 0.008、75As 0.02、137Ba 0.03、9Be 0.02、111Cd 0.02、52Cr 0.04、59Co 0.002、63Cu 0.004、206+207+208Pb 0.015、55Mn 0.007、202Hg 0.2、98Mo 0.005、 60Ni 0.07、82Se 1.3、107Ag 0.004、205Tl 0.014、232Th 0.005、238U 0.005、51V 0.006、 66Zn 0.07),《GB/T 8538-2008饮用天然矿泉水检验方法》(暨《GB/T 5750-2006》)提出 31个元素( 107Ag 0.03、27Al 0.6、75As 0.09、11B 0.9、135Ba 0.3、9Be 0.03、40Ca 6.0、111Cd 0.06、59Co 0.03、52Cr 0.09、63Cu 0.09、56Fe 0.9、 39K 3.0、7Li 0.3、24Mg 0.4, 55Mn 0.06、98Mo 0.06、23Na 7.0、60Ni 0.07、208Pb 0.07、121Sb 0.07、77Se 0.09、88Sr 0.09、118Sn 0.09、232Th 0.06、203Tl 0.01, 48Ti 0.04、235U 0.04、51V0.07、66Zn 0.8、202Hg0.07),我们利用 PQ ExCell ICP-MS自二○○二年以来进行了检测饮用水中微量元素方法研究,提出了 15个元素(7Li 0.051、27Al 1.8、51V 0.044、52Cr 0.23、55Mn 0.20、59Co 0.17、60Ni 0.99、 65Cu 0.17、66Zn 0.42、82Sr 0.052、98Mo 0.037、107Ag 0.013、114Cd 0.022、137Ba 0.17、208Pb 0.088),现在仪器更换为 XSeries2后在性能方面发生很多变化,根据分析测试需要在检测项目方面也发生了很多变化,需要通过方法研究解决这些变化,另外在《 EPA200.8》和《GB/T 8538-2008》之间也存在诸多不同之处,需要通过研究进行确认。在国内已有很多 ICP-MS检测水中微量元素的文献发表,检测元素最多的已经达到 44项,检测方法存在很多不同的地方,对于这些文献提出的结论,是否值得我们借鉴,需要通过研究和实验进行验证。 6.1多元素的检测条件研究
6.1.1在 XSeries2仪器能够检测的元素
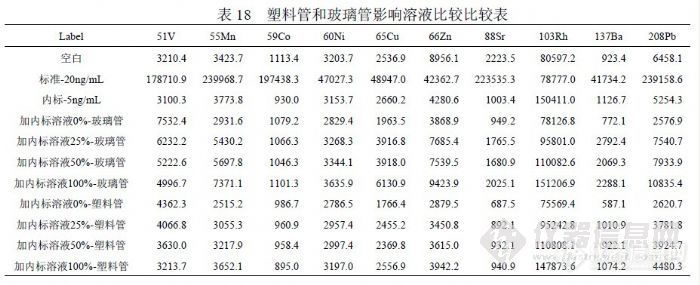
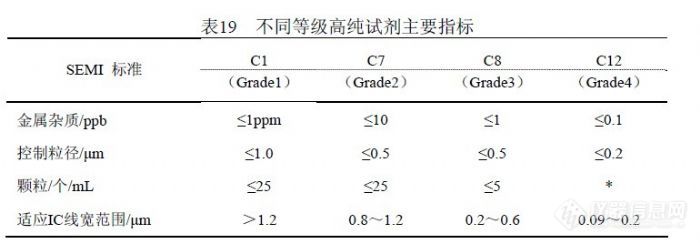
在理论上 XSeries2 ICP-MS仪器能够分析很多元素,但具体日常水分析能够检测的元素就要慎重确定,我们为此确定两个原则,其一是被检测元素的干扰已经明确并能够校正,其二是检出限能够满足有关标准的要求。在过去的工作中,通过实验和研究确定当前能够检测的元素有: Li、Be、Al、V、 Cr、Mn、Co、Ni、Cu、Zn、As、Se、Sr、Mo、Ag、Cd、I、Ba、Pb、Fe、 Hg和 Rh、Au。其它元素,由于干扰、背景、灵敏度、精密度等具体情况没有明确,不能做为定量元素进行检测。 6.1.2 检测溶液酸度的选择进行 ICP-MS分析的介质一般都选用“ 1%的硝酸”,我们的实验结果也支持这一点,《EPA200.8》和《GB/T 8538-2008》的分析介质也都选择“ 1%的硝酸”。我们在日常分析过程中,观察到当标准溶液或样品浓度超过 20ng/mL时,1%硝酸需要较长的清洗时间,通常超过 3min,否则将导致交叉污染。我们把溶液酸度变成“ 2%的硝酸”后,清洗时间缩短到 50s之内,能够提高分析效率。ICP-MS分析要求溶液酸度不能超过 5%。6.1.3 内标的选择我们考虑到如果样品中含有内标元素,会严重干扰内标对分析结果的校正;如果为抵消这种干扰而提高内标元素的浓度,则由于内标信号值过高而失去对低信号值的校正作用。通过实验确定在线加入浓度为 5ng/mL的 Rh(铑)作为内标,取得较好效果。内标信号值约 150000cps,对测试过程中仪器条件变化产生的影响能够有效补偿。内标溶液在加入样品溶液和标准溶液时,有可能定量引入目标物。如果样品带有空白,通过扣除空白值能够消除其影响;直接检测的样品没有空白,则需要通过使用标准加入法确定内标溶液所含目标物的浓度,然后予以扣除。内标溶液含有目标物的主要原因有几个,其一配制内标的纯水纯度影响,即使 18.3MΩ以上纯水仍含有一定量目标物,能够对低浓度目标物产生影响;其二配制内标的酸会含有目标物,高纯酸影响相对较小;其三内标元素的单标溶液背景纯度不够会含有目标物;其四配制过程中环境悬浮颗粒进入内标溶液会引入目标物;其五检测过程中环境悬浮颗粒进入内标溶液会引入目标物。为保证 ICP-MS检测水平和数据可靠,从纯水制备到样品准备再到样品检测,均应在洁净环境中完成,洁净环境的洁净度能够决定检测所能达到的水平。 6.1.4 检测溶液盛装容器的选择表 18是验证内标溶液对检测结果影响的实验,但观察到玻璃管导致下列元素的背景明显高于塑料管,并且玻璃管更容易污染且不容易清洗干净。除了标准溶液,在玻璃管和塑料管内做相同实验进行比较,按顺序是:空白溶液、加入 25%(体积比)内标溶液、加入 50%内标溶液、内标溶液,然后同标准溶液一样上机测试。统计结果表明,扣除加入内标溶液所带入信号值,玻璃管溶液背景明显高于塑料管,并且玻璃管之间出现污染情况。![]() 在 Hg的实验过程中发现塑料容器对 Hg有吸附影响,足以改变分析结果的准确度和精密度。根据出现的这种情况,Hg的检测宜单独采用玻璃容量瓶做为容器,而其它元素应使用 PP、PE或 PET塑料容器。6.1.5 检测过程中环境的影响因素在 ICP-MS分析中,氩气的纯度至关重要,国内氩气供应存在较多不确定因素。我们经历过因氩气纯度不足曾导致下列情况出现,其一仪器不能点火,其二仪器反射功率过大使灵敏度和精密度下降,其三是 Kr背景过高对 Se、As的检测产生严重干扰并无法消除,因为干扰信号达到( 6000~30000) cps已经远远超过样品检测信号。水的纯度直接影响检测结果,定期维护纯水设备是保证纯水纯度的必要保证,但还有其它因素能够影响纯水的纯度。环境粉尘和空气微生物能够进入纯水中造成污染,粉尘含有目标物造成纯水背景偏高,严重可能会湮没检测信号,微生物使有机质含量升高,能够对检测信号形成抑制。所以有必要在洁净室中制备 ICP-MS分析所需纯水。国产优级纯硝酸不适合 ICP-MS分析,尤其是浓度较低的目标物检测。用于分析测试的高纯酸要求杂质浓度控制在“ 0.1ng/mL”以内,这样酸度在(1~2)%时杂质产生的背景浓度将在(0.001~0.002)ng/mL,能够不对 0.0x ng/mL级别的目标物产生干扰,这也就是纯水纯度、硝酸纯度、环境水平决定分析测试水平的原因,而不单单是仪器性能决定分析测试水平。用于 ICP-OES检测的硝酸要求杂质控制在“ 1.0ng/mL”以内,而用于石墨炉原子吸收的硝酸或盐酸要求杂质控制在“10.0ng/mL”以内,而国产优级纯硝酸杂质常常超过“100 ng/mL”。超净高纯试剂( Ultra-clean and High-purity Reagents)在国际上通称为工艺化学品(Process Chemicals),美欧和中国台湾地区又称湿化学品(Wet Chemicals),是超大规模集成电路制作过程中的关键性基础化工材料之一,具有品种多、用量大、技术要求高、贮存有效期短和腐蚀性强等特点。 1975 年,国际半导体设备与材料组织(SEMI)制定了国际统一的超净高纯试剂标准,如表 19所示。国际上制备 SEMI-C1 到 SEMI-C12 级超净高纯试剂的技术都已经趋于成熟。国内超净高纯试剂的研发水平及生产技术水平与国际上的先进技术水平相比尚有一定的差距,目前 5μmIC技术用 MOS 级试剂的生产技术已经成熟,并已转化为规模生产。0.8~1.2μm 技术用超净高纯试剂(相当于国际 SEMI 标准 C7水平)的产业化技术基本成熟,初步形成生产规模。0.2~0.6μm 技术用超净高纯试剂(相当于国际 SEMI标准 C8水平)的工艺制备技术及分析测试技术有所突破,但由于受相关配套条件的制约,产业化技术还有待进一步的完善。我们目前能够获得的最好的高纯试剂就是北京化学试剂研究所的 BV-III(C7)试剂。
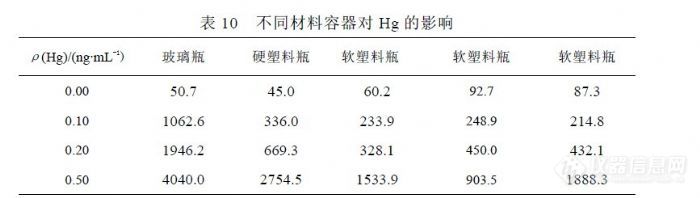
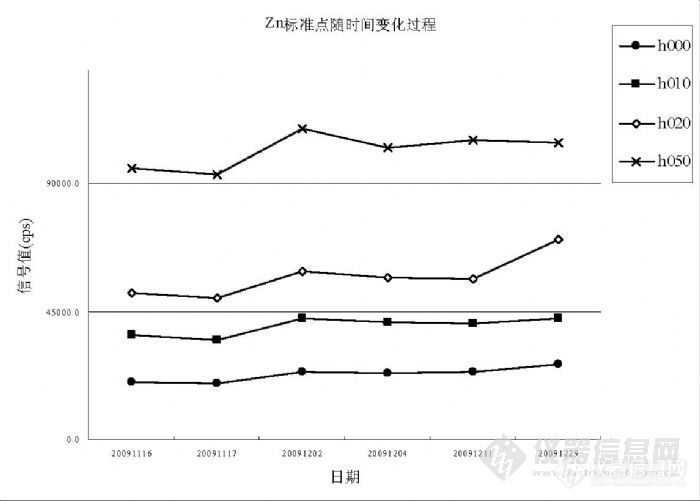
在 Hg的实验过程中发现塑料容器对 Hg有吸附影响,足以改变分析结果的准确度和精密度。根据出现的这种情况,Hg的检测宜单独采用玻璃容量瓶做为容器,而其它元素应使用 PP、PE或 PET塑料容器。6.1.5 检测过程中环境的影响因素在 ICP-MS分析中,氩气的纯度至关重要,国内氩气供应存在较多不确定因素。我们经历过因氩气纯度不足曾导致下列情况出现,其一仪器不能点火,其二仪器反射功率过大使灵敏度和精密度下降,其三是 Kr背景过高对 Se、As的检测产生严重干扰并无法消除,因为干扰信号达到( 6000~30000) cps已经远远超过样品检测信号。水的纯度直接影响检测结果,定期维护纯水设备是保证纯水纯度的必要保证,但还有其它因素能够影响纯水的纯度。环境粉尘和空气微生物能够进入纯水中造成污染,粉尘含有目标物造成纯水背景偏高,严重可能会湮没检测信号,微生物使有机质含量升高,能够对检测信号形成抑制。所以有必要在洁净室中制备 ICP-MS分析所需纯水。国产优级纯硝酸不适合 ICP-MS分析,尤其是浓度较低的目标物检测。用于分析测试的高纯酸要求杂质浓度控制在“ 0.1ng/mL”以内,这样酸度在(1~2)%时杂质产生的背景浓度将在(0.001~0.002)ng/mL,能够不对 0.0x ng/mL级别的目标物产生干扰,这也就是纯水纯度、硝酸纯度、环境水平决定分析测试水平的原因,而不单单是仪器性能决定分析测试水平。用于 ICP-OES检测的硝酸要求杂质控制在“ 1.0ng/mL”以内,而用于石墨炉原子吸收的硝酸或盐酸要求杂质控制在“10.0ng/mL”以内,而国产优级纯硝酸杂质常常超过“100 ng/mL”。超净高纯试剂( Ultra-clean and High-purity Reagents)在国际上通称为工艺化学品(Process Chemicals),美欧和中国台湾地区又称湿化学品(Wet Chemicals),是超大规模集成电路制作过程中的关键性基础化工材料之一,具有品种多、用量大、技术要求高、贮存有效期短和腐蚀性强等特点。 1975 年,国际半导体设备与材料组织(SEMI)制定了国际统一的超净高纯试剂标准,如表 19所示。国际上制备 SEMI-C1 到 SEMI-C12 级超净高纯试剂的技术都已经趋于成熟。国内超净高纯试剂的研发水平及生产技术水平与国际上的先进技术水平相比尚有一定的差距,目前 5μmIC技术用 MOS 级试剂的生产技术已经成熟,并已转化为规模生产。0.8~1.2μm 技术用超净高纯试剂(相当于国际 SEMI 标准 C7水平)的产业化技术基本成熟,初步形成生产规模。0.2~0.6μm 技术用超净高纯试剂(相当于国际 SEMI标准 C8水平)的工艺制备技术及分析测试技术有所突破,但由于受相关配套条件的制约,产业化技术还有待进一步的完善。我们目前能够获得的最好的高纯试剂就是北京化学试剂研究所的 BV-III(C7)试剂。![]() 仪器工作环境中粉尘能够进入标准溶液并形成积累,积累到一定程度将改变标准曲线,因此标准溶液即使没有使用完也应定期更换,更换周期不能超过一个月,而标准溶液使用超过一半时必须更换,因为随溶液体积减少被污染的程度将成倍数增加。下图是 Zn标准溶液各点 h000(0.0ng/mL)、h010(10.0ng/mL)、h020(20.0ng/mL)、h050(50.0ng/mL)从 11月 15日配制后到 12月 29日的变化过程。除 20091202因调节分辨率而信号产生波动外,其余各点均在相同分辨率条件下进行检测,相同浓度点的信号值呈上升趋势。 H020的 20091229浓度点就是因瓶盖儿没有拧紧,放置约一个星期时间后 Zn受到污染,信号值升高幅度较大使标准曲线线性被破坏。观察期间仪器工作间温度保持在 20℃,湿度 35%,洁净工作间按 100000级设计。标准溶液定容体积 50.0mL,在 12月 29日剩余体积均在 40mL左右,信号值变化与溶液体积变化没有关联,因为存在明显变化的情况仅发生在 Zn、Cu、Cr、Pb等几个元素上,以 Zn最显著,其它元素没有参与这种变化趋势。
仪器工作环境中粉尘能够进入标准溶液并形成积累,积累到一定程度将改变标准曲线,因此标准溶液即使没有使用完也应定期更换,更换周期不能超过一个月,而标准溶液使用超过一半时必须更换,因为随溶液体积减少被污染的程度将成倍数增加。下图是 Zn标准溶液各点 h000(0.0ng/mL)、h010(10.0ng/mL)、h020(20.0ng/mL)、h050(50.0ng/mL)从 11月 15日配制后到 12月 29日的变化过程。除 20091202因调节分辨率而信号产生波动外,其余各点均在相同分辨率条件下进行检测,相同浓度点的信号值呈上升趋势。 H020的 20091229浓度点就是因瓶盖儿没有拧紧,放置约一个星期时间后 Zn受到污染,信号值升高幅度较大使标准曲线线性被破坏。观察期间仪器工作间温度保持在 20℃,湿度 35%,洁净工作间按 100000级设计。标准溶液定容体积 50.0mL,在 12月 29日剩余体积均在 40mL左右,信号值变化与溶液体积变化没有关联,因为存在明显变化的情况仅发生在 Zn、Cu、Cr、Pb等几个元素上,以 Zn最显著,其它元素没有参与这种变化趋势。 ![]()
图 14标准溶液受环境影响变化曲线
6.1.6 饮用水中稀土元素直接检测的研究我们通过实验研究了饮用水中稀土元素的直接测试,配制标准曲线的浓度范围在(0.0x~1xx.0)ng/mL之间,检测信号值在(1xxx~xxxxxx)cps之间。从标准曲线的相关系数、斜率、检出限和检测限等参数情况判断,直接检测稀土元素具有可行性;从实际样品检测情况分析,还存在疑问,因为只有 Y、La、Ce的多数样品信号值超过 10000cps,Pr、Nd的多数样品信号值能够超过 1000cps,其余信号值都在 1000cps以下,重稀土元素很多样品信号值甚至在 100cps以下,这样使检测的精密度不能保证。经过测算,当信号值<100cps时,RSD在(22.46~61.22)%范围,平均 38.48%;当信号值在( 100~1000)cps之间时,RSD在(0.84~23.16)%范围,平均 8.36%;当信号值在(1000~10000)cps之间时,RSD在(0.76~8.67)%范围,平均 3.68%。从检测精密度方面分析,水中稀土元素之间检测需要灵敏度更高的仪器和更好的环境条件(洁净度在 1000级以上)。![]() 6.2各元素的灵敏度和相互关系研究
6.2各元素的灵敏度和相互关系研究
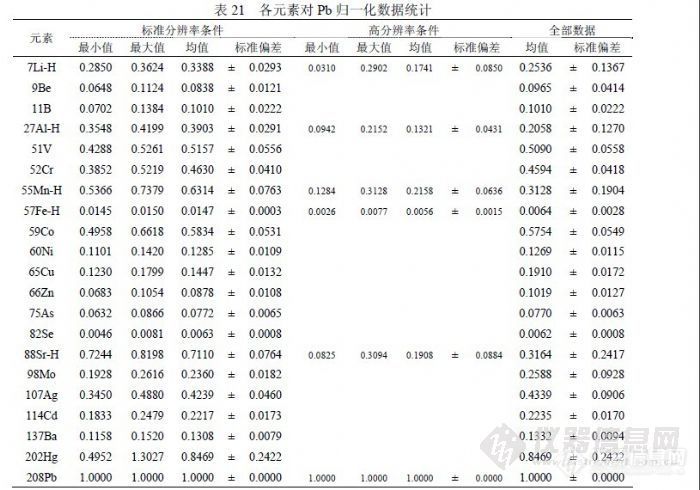
6.2.1影响检测元素灵敏度的因素在 ICP-MS检测中,影响灵敏度的因素有多种,但能够影响各元素之间灵敏度变化的因素就不多了。离子透镜电位变化可以改变轻、重质量数的灵敏度,雾化气流量变化能够通过“质量歧视”效应改变轻、重质量数的灵敏度,但这两种因素在相对固定的周期内保持不变。分辨率是影响各元素灵敏度高低和各元素之间灵敏度变化的主要因素,通常情况下,控制分辨率的 Be、Co、In、U四个调节电位参数一但确定下来也会较长时间内保持不变,但这四个电位参数会受到环境湿度影响产生一定变化,必要时需要做微调。分辨率与灵敏度的关系主要是,随分辨率提高灵敏度将下降。在检测中观察到,分辨率确定不变的情况下,各元素之间的灵敏度能够保持固定的比例关系。如果某个元素单独选择高分辨率状态,其与其它元素原有的比例关系将发生变化。 6.2.2 不稳定元素与相邻元素之间的关系为了了解各元素灵敏度变化趋势,我们统计了近一年多标准曲线的检测数据,包括在 PQ ExCell和 XSeries2仪器的检测数据,时间跨五个季节,不同时间配制的相同浓度的标准曲线数据,同一标准曲线溶液在不同时间进行检测的数据,不同的有证书标准溶液所配制的不同标准曲线检测数据等。为了能够在相同条件下比较数据变化趋势,在每批检测数据中以 Pb为统计基点,其它元素对 Pb做归一化处理统计出用于比较的参数。![]() 表中带“ -H”后缀的元素是需要采用高分辩率方式检测的,“全部数据”栏是各种情况数据包括异常数据全部进行统计的结果。表中数据显示,在标准分辨率模式下各元素间灵敏度能够保持固定比例关系,相对标准偏差在(5~30)%之间,轻质量数元素变化相对大,易变化的元素如 Hg、Ag等变化相对也大,多数元素相对标准偏差在 10%左右;高分辩率模式的数据相对较差,这是因为采用高分辩率后峰宽变窄(标准分辨率峰宽 0.70amu,高分辩率峰宽 0.35amu),信号灵敏度下降,精密度相应降低,但有利于检测高浓度样品,而采用高分辩率模式的都是样品中含量比较高的元素。如果以Be、Co为基点分别统计轻质量数元素、中质量数元素,由于元素间行为变化趋于一致,能够使元素间灵敏度比例关系变化程度更小,相对标准偏差在 10%以内。以 Mn为基点单独统计高分辩率模式参数,相对标准偏差能够控制在 20%以内。统计元素间灵敏度变化关系可以用于以下几个方面,其一检查测试过程中标准曲线的配制是否存在问题,其二判断样品检测结果是否存在异常情况,其三可以通过扫描方式判断未知样品中目标物含量高低,其四在缺乏标准品的情况下进行半定量检测。 6.2.3 如何使用半定量方法 XSeries2的 PlasmaLab软件有半定量功能,利用各元素的理论灵敏度曲线估算浓度。其方法是通过定量元素的标准溶液确定由测量值向理论值校正的幅度,然后用这个参数去修正样品信号值,最后以灵敏度曲线为依据估算样品含量。灵敏度曲线的绘制与定量元素的质量数( m/z)、相对灵敏度系数(RSF)、丰度(Abundance)有关。但 PlasmaLab软件进行数据计算时存在缺陷,就是在完成标准溶液检测后会统计出标准曲线的截距,然后在任何样品信号的修正中扣除这个截距,这个截距主要受标准溶液的背景影响。由于水分析有一定特殊性,直接进样检测的样品实际没有空白,往往在扣除截距后信号值已成负值,偏离真实结果,浓度低的样品受此影响比较显著;如果样品带空白,可以通过扣空白抵消截距影响。所以为保证检测结果可靠性,需要使用没有修正的数据脱机另行统计。使用半定量方法估算分析结果的情况出现在暂时缺乏标准、含量过高不宜直接定量、对分析结果要求不高等情况,使用仪器的预扫描功能获取样品信息,利用元素间灵敏度的相关关系估算结果。 6.2.4 定量方法配制标准溶液,使用单元素标准储备液配制混合标准溶液 A1,包含元素 Li、Be、Al、V、Cr、Mn、Co、Ni、Cu、Zn、As、Se、Sr、Mo、Cd、 Ba、Pb、Fe等,浓度 ρ(M)=1.0μg/mL;使用单元素标准储备液配制混合标准溶液 A2,包含元素 Li、Al、Mn、Fe、Sr等,浓度 ρ(M)=10.0μg/mL;使用单元素标准储备液配制标准溶液 A3,包含元素 Ag等,溶液需要避光保存,浓度 ρ(M)=1.0μg/mL;使用单元素标准储备液配制标准溶液 A4,包含元素 Hg等,浓度 ρ(M)=1.0μg/mL;使用单元素标准储备液配制标准溶液 A5,包含元素 I等,浓度 ρ(M)=1.0μg/mL。直接使用的标准溶液 A6(ICPMS-43,编号 021809),包含元素 As、Cd、Cr、Cu、Fe、Pb、Mn、Ni、Se、V、Zn、 Be、Cs、Ce、Mg、Sr、Tl、Dy、Er、Ca、Ag、Al、B、Ba、K、Na、Eu、 Ga、Gd、Ho、La、Lu、Nd、P、S、Pr、Ru、Sm、Th、Tm、U、Yb等,浓度 ρ(M)=(10.0±0.028)μg/mL;直接使用的标准溶液 A7(GBW(E)080670,样品编号 0904),包含元素 K、Na、Ca、Mg、Fe、Li、Al、V、Cr、Mn、 Co、Ni、Cu、Zn、As、Se、Sr、Ag、Cd、Ba、Bi、Pb、Be、Cs、Rb等,浓度 ρ(M)=(10.0±0.03)μg/mL。配制标准曲线,使用 A1配制浓度为(0.0、1.0、10.0、100.0)ng/mL的系列 B1;使用 A2配制(0.0、500.0、1000.0)ng/mL的系列 B2;使用 A3配制浓度为(0.0、1.0、10.0、100.0)ng/mL的系列 B3;使用 A4配制(0.0、1.0、10.0、 100.0)ng/mL的系列 B4;使用 A5配制浓度为(0.0、1.0、10.0、100.0)ng/mL的系列 B5;使用 A7配制(0.0、10.0、40.0、100.0)ng/mL的系列 H1;使用 A6配制(0.0、10.0)ng/mL的监控溶液 JK。选择同位素 7Li、9Be、27Al、51V、52Cr、55Mn、57Fe、59Co、60Ni、 65Cu、66Zn、75As、82Se、88Sr、98Mo、107Ag、114Cd、127I、137Ba、 202Hg、208Pb进行定量测试,选择 103Rh为内标元素,内标浓度 5ng/mL(含 200ng/mL的 Au)。定量测试选择“跳峰”方式,部分元素 7Li、27Al、 55Mn、57Fe、88Sr选择高分辩率模式,参加确定灵敏度曲线的元素选择 7Li、 59Co、66Zn、82Se、98Mo、114Cd、137Ba、208Pb。对于饮用矿泉水样品、生活饮用水样品可以直接定量检测,主运行 3次,其它样品不可以直接定量测试,应运行预扫描半定量观察含量高低,再确定稀释、其它处理或直接检测。因为仪器软件处理数据时在样品信号中扣除包括系列空白信息的标准曲线截距,与样品检测实际情况有矛盾,所以不能使用仪器软件中“校正浓度”和“校正信号”页面的信息用于检测报告内容,应复制“未校正信号”页面的内容到“电子表格”中进行手工处理。处理原则是,各系列和标准在分别扣除自己的空白后参与计算,直接检测的样品不扣空白(如果随样品带有空白应予扣除),经过处理的样品必须扣除“过程空白”,直接检测的样品应考虑扣除在线内标引入的背景。在线内标所含目标物背景值正常情况下应通过标准加入法准确测定,然后在样品结果中扣除。检测报告应包含如下内容:分析批号、分析编号、分析项目、分析技术、依据标准、设备编号、仪器条件(经常变化并能够影响检测结果和仪器基本状态)、环境温湿度、标准曲线(信号值、浓度值、相关系数、斜率、截距)、样品结果(信号值、含量)、计算公式、分析日期、页码(页序、页数)、源文件信息(名称、路径)、分析人和校核人的签字(每页检测报告必须有这两个签字)、首页还要有负责人和组长签字。 6.3 检出限检出限按(4.4.2)方法统计,但检测限是按 10倍标准偏差进行统计,列出的“国标检出限”是指《GB/T 8538-2008》有关 ICP-MS检测部分提出的参数。
表中带“ -H”后缀的元素是需要采用高分辩率方式检测的,“全部数据”栏是各种情况数据包括异常数据全部进行统计的结果。表中数据显示,在标准分辨率模式下各元素间灵敏度能够保持固定比例关系,相对标准偏差在(5~30)%之间,轻质量数元素变化相对大,易变化的元素如 Hg、Ag等变化相对也大,多数元素相对标准偏差在 10%左右;高分辩率模式的数据相对较差,这是因为采用高分辩率后峰宽变窄(标准分辨率峰宽 0.70amu,高分辩率峰宽 0.35amu),信号灵敏度下降,精密度相应降低,但有利于检测高浓度样品,而采用高分辩率模式的都是样品中含量比较高的元素。如果以Be、Co为基点分别统计轻质量数元素、中质量数元素,由于元素间行为变化趋于一致,能够使元素间灵敏度比例关系变化程度更小,相对标准偏差在 10%以内。以 Mn为基点单独统计高分辩率模式参数,相对标准偏差能够控制在 20%以内。统计元素间灵敏度变化关系可以用于以下几个方面,其一检查测试过程中标准曲线的配制是否存在问题,其二判断样品检测结果是否存在异常情况,其三可以通过扫描方式判断未知样品中目标物含量高低,其四在缺乏标准品的情况下进行半定量检测。 6.2.3 如何使用半定量方法 XSeries2的 PlasmaLab软件有半定量功能,利用各元素的理论灵敏度曲线估算浓度。其方法是通过定量元素的标准溶液确定由测量值向理论值校正的幅度,然后用这个参数去修正样品信号值,最后以灵敏度曲线为依据估算样品含量。灵敏度曲线的绘制与定量元素的质量数( m/z)、相对灵敏度系数(RSF)、丰度(Abundance)有关。但 PlasmaLab软件进行数据计算时存在缺陷,就是在完成标准溶液检测后会统计出标准曲线的截距,然后在任何样品信号的修正中扣除这个截距,这个截距主要受标准溶液的背景影响。由于水分析有一定特殊性,直接进样检测的样品实际没有空白,往往在扣除截距后信号值已成负值,偏离真实结果,浓度低的样品受此影响比较显著;如果样品带空白,可以通过扣空白抵消截距影响。所以为保证检测结果可靠性,需要使用没有修正的数据脱机另行统计。使用半定量方法估算分析结果的情况出现在暂时缺乏标准、含量过高不宜直接定量、对分析结果要求不高等情况,使用仪器的预扫描功能获取样品信息,利用元素间灵敏度的相关关系估算结果。 6.2.4 定量方法配制标准溶液,使用单元素标准储备液配制混合标准溶液 A1,包含元素 Li、Be、Al、V、Cr、Mn、Co、Ni、Cu、Zn、As、Se、Sr、Mo、Cd、 Ba、Pb、Fe等,浓度 ρ(M)=1.0μg/mL;使用单元素标准储备液配制混合标准溶液 A2,包含元素 Li、Al、Mn、Fe、Sr等,浓度 ρ(M)=10.0μg/mL;使用单元素标准储备液配制标准溶液 A3,包含元素 Ag等,溶液需要避光保存,浓度 ρ(M)=1.0μg/mL;使用单元素标准储备液配制标准溶液 A4,包含元素 Hg等,浓度 ρ(M)=1.0μg/mL;使用单元素标准储备液配制标准溶液 A5,包含元素 I等,浓度 ρ(M)=1.0μg/mL。直接使用的标准溶液 A6(ICPMS-43,编号 021809),包含元素 As、Cd、Cr、Cu、Fe、Pb、Mn、Ni、Se、V、Zn、 Be、Cs、Ce、Mg、Sr、Tl、Dy、Er、Ca、Ag、Al、B、Ba、K、Na、Eu、 Ga、Gd、Ho、La、Lu、Nd、P、S、Pr、Ru、Sm、Th、Tm、U、Yb等,浓度 ρ(M)=(10.0±0.028)μg/mL;直接使用的标准溶液 A7(GBW(E)080670,样品编号 0904),包含元素 K、Na、Ca、Mg、Fe、Li、Al、V、Cr、Mn、 Co、Ni、Cu、Zn、As、Se、Sr、Ag、Cd、Ba、Bi、Pb、Be、Cs、Rb等,浓度 ρ(M)=(10.0±0.03)μg/mL。配制标准曲线,使用 A1配制浓度为(0.0、1.0、10.0、100.0)ng/mL的系列 B1;使用 A2配制(0.0、500.0、1000.0)ng/mL的系列 B2;使用 A3配制浓度为(0.0、1.0、10.0、100.0)ng/mL的系列 B3;使用 A4配制(0.0、1.0、10.0、 100.0)ng/mL的系列 B4;使用 A5配制浓度为(0.0、1.0、10.0、100.0)ng/mL的系列 B5;使用 A7配制(0.0、10.0、40.0、100.0)ng/mL的系列 H1;使用 A6配制(0.0、10.0)ng/mL的监控溶液 JK。选择同位素 7Li、9Be、27Al、51V、52Cr、55Mn、57Fe、59Co、60Ni、 65Cu、66Zn、75As、82Se、88Sr、98Mo、107Ag、114Cd、127I、137Ba、 202Hg、208Pb进行定量测试,选择 103Rh为内标元素,内标浓度 5ng/mL(含 200ng/mL的 Au)。定量测试选择“跳峰”方式,部分元素 7Li、27Al、 55Mn、57Fe、88Sr选择高分辩率模式,参加确定灵敏度曲线的元素选择 7Li、 59Co、66Zn、82Se、98Mo、114Cd、137Ba、208Pb。对于饮用矿泉水样品、生活饮用水样品可以直接定量检测,主运行 3次,其它样品不可以直接定量测试,应运行预扫描半定量观察含量高低,再确定稀释、其它处理或直接检测。因为仪器软件处理数据时在样品信号中扣除包括系列空白信息的标准曲线截距,与样品检测实际情况有矛盾,所以不能使用仪器软件中“校正浓度”和“校正信号”页面的信息用于检测报告内容,应复制“未校正信号”页面的内容到“电子表格”中进行手工处理。处理原则是,各系列和标准在分别扣除自己的空白后参与计算,直接检测的样品不扣空白(如果随样品带有空白应予扣除),经过处理的样品必须扣除“过程空白”,直接检测的样品应考虑扣除在线内标引入的背景。在线内标所含目标物背景值正常情况下应通过标准加入法准确测定,然后在样品结果中扣除。检测报告应包含如下内容:分析批号、分析编号、分析项目、分析技术、依据标准、设备编号、仪器条件(经常变化并能够影响检测结果和仪器基本状态)、环境温湿度、标准曲线(信号值、浓度值、相关系数、斜率、截距)、样品结果(信号值、含量)、计算公式、分析日期、页码(页序、页数)、源文件信息(名称、路径)、分析人和校核人的签字(每页检测报告必须有这两个签字)、首页还要有负责人和组长签字。 6.3 检出限检出限按(4.4.2)方法统计,但检测限是按 10倍标准偏差进行统计,列出的“国标检出限”是指《GB/T 8538-2008》有关 ICP-MS检测部分提出的参数。![]() 在方法研究中了解到,检出限由标准偏差和标准曲线斜率组成,能够影响斜率的主要因素是仪器的维护水平和仪器的调试水平,因为仪器在日常检测过程中不可能总是处于最佳状态,一般都以满足本次测试需要为目的,如果调试仪器的水平较高可以使仪器在较好的状态下工作,通过炬箱、离子透镜、碰撞池、四极杆、检测器等参数组合获得较高的灵敏度和稳定性,与低水平调试相比有时会形成天壤之别,而仪器维护主要包含仪器硬件所处于的工作状态,对灵敏度和稳定性有影响的硬件主要有进样管是否顺畅或泄漏、雾化器是否完好、采样锥和截取锥是否清洗、排风风量是否正常、机械泵抽真空是否正常其泵油是否按时更换、真空腔是否受到污染、雾室和炬管是否受到污染等,只要有一项出现问题就会使仪器灵敏度或稳定性不同程度的下降;如前面所述仪器不正常势必影响标准偏差变差,若仪器处于正常状态,能够影响标准偏差的主要因素就是空白溶液制备水平,包含纯水纯度、硝酸纯度、容器洁净度和环境干扰强弱等因子。在研究过程中,使用不同方法统计检出限参数进行比较,以确定水分析中哪些同位素被何因子影响。
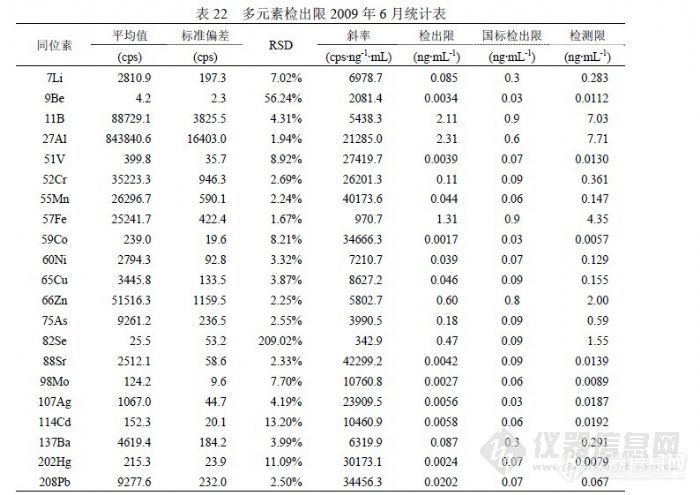
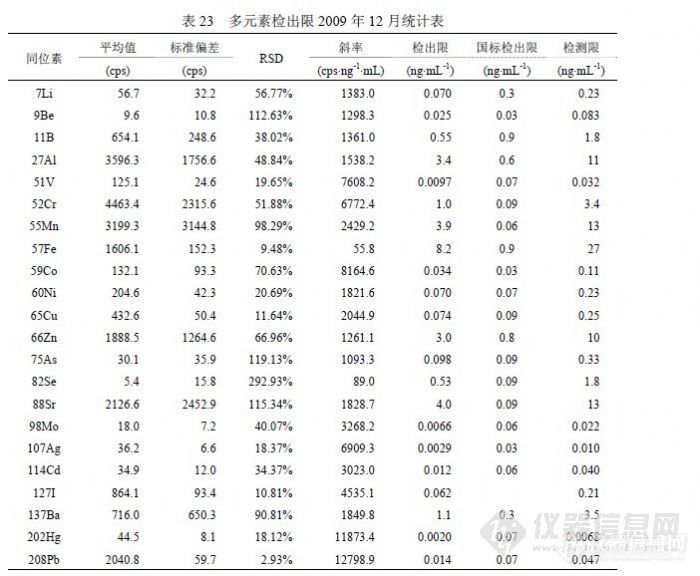
在方法研究中了解到,检出限由标准偏差和标准曲线斜率组成,能够影响斜率的主要因素是仪器的维护水平和仪器的调试水平,因为仪器在日常检测过程中不可能总是处于最佳状态,一般都以满足本次测试需要为目的,如果调试仪器的水平较高可以使仪器在较好的状态下工作,通过炬箱、离子透镜、碰撞池、四极杆、检测器等参数组合获得较高的灵敏度和稳定性,与低水平调试相比有时会形成天壤之别,而仪器维护主要包含仪器硬件所处于的工作状态,对灵敏度和稳定性有影响的硬件主要有进样管是否顺畅或泄漏、雾化器是否完好、采样锥和截取锥是否清洗、排风风量是否正常、机械泵抽真空是否正常其泵油是否按时更换、真空腔是否受到污染、雾室和炬管是否受到污染等,只要有一项出现问题就会使仪器灵敏度或稳定性不同程度的下降;如前面所述仪器不正常势必影响标准偏差变差,若仪器处于正常状态,能够影响标准偏差的主要因素就是空白溶液制备水平,包含纯水纯度、硝酸纯度、容器洁净度和环境干扰强弱等因子。在研究过程中,使用不同方法统计检出限参数进行比较,以确定水分析中哪些同位素被何因子影响。![]() 表 22是 2009年 6月实验结果,使用一个空白溶液进行 12次进样统计的检出限参数,正常情况下相当于仪器检出限。与《 GB/T 8538-2008》提出的参数比较,202Hg、137Ba、114Cd、107Ag、98Mo、88Sr、59Co、60Ni、 51V、9Be、7Li等同位素远低于国标,它们在一般矿物土壤中含量很低,且容易清洗不易污染仪器背景,同时这些同位素也具有相对足够高的灵敏度; 52Cr、55Mn、65Cu、66Zn、208Pb等同位素与国标接近,它们的特点是空白信号值较大但 RSD较好,应受纯水、硝酸影响的较大; 11B、27Al、75As的精密度都很好,但由于空白信号值过大导致检出限参数不理想,其原因主要是制备空白溶液的纯水所含杂质含量过高,同时受环境因素影响,可能空气中粉尘进入溶液产生污染;57Fe、82Se是检测同位素中灵敏度比较低的两个,57Fe的空白信号值较大,主要的影响因素是纯水和硝酸,若检测 10ng/mL以下浓度的 Fe需要纯度等级更高的纯水、硝酸及 1000级(或更好)洁净条件,82Se的检测精密度受到 83Kr干扰,改善的方法就是提高氩气纯度,通过验证若不扣除 83Kr的干扰检出限反而会好,因为信号值增加改善了精密度,但能够使低浓度样品分析结果偏高至少一个数量级。有观点认为空气中粉尘影响 ICP-MS的检测结果不可理解,这是因为在 ICP-MS技术应用之前原子吸收所检测浓度相对较高,感受不到环境粉尘的影响,实际上微电子行业的实验室在其制备、检测环境都达到 1000级洁净,更高的达到 100级洁净,在试剂纯度方面采用 SEMI国际标准按 C1~C12级,最差的 C1级相当于优级纯。其有关资料认为影响检测结果的沾污一般有四类:颗粒(来自设备、超净间空气、工艺气体和化学试剂、去离子水),金属(来自设备、超净高纯试剂、离子注入、灰化、反应离子刻蚀),有机物(来自超净间气体、光刻胶残渣、贮存容器、工艺化学试剂),自然氧化物(来自超净间湿度、去离子水冲洗)。我们在实验中也观察到,暴露空气的空白溶液随放置时间增加,元素 Pb、Ba、Cd、Zn、Cu、Ni、B、Cr等不同程度随之浓度升高,另外内标也含有检测元素,并且不同时期配制的内标溶液含有干扰物的浓度不同。经常出现的情况是,某些元素的空白信号值中有很大部分来自在线内标溶液,如 27Al有 70%、11B有 70%、51V有 40%、55Mn有 50%、57Fe有 50%、59Co有 50%、60Ni有 80%、65Cu有 70%、66Zn有 90%、82Sr有 50%、127I有 60%、137Ba有 50%等。表 22的仪器条件处于灵敏度较高时期,其内标信号可达 30万,后出于延长检测器寿命考虑取下屏蔽圈,灵敏度降为 30%。表 23是 12月以 12份独立空白溶液检测后统计的检出限参数,每份溶液均 1次进样连续采集 3次取均值参加统计。与表 22相比,能够确定 6月实验的空白溶液中 Li、B、As、Ag受到污染,因为表 23的空白信号偏低幅度与灵敏度下降幅度不符,而且这 4个同位素尽管精密度不及表 22,但获得了更好的检出限参数。同位素 137Ba、114Cd、98Mo、88Sr、66Zn、65Cu、 60Ni、59Co、57Fe、55Mn、52Cr、51V等都因精密度变差而获得比表 22更差的检出限参数,27Al的原因类似,其空白信号虽有效下降但没有抵消精密度变差的效果。9Be因灵敏度下降而导致检出限上升,208Pb和 202Hg的检出限水平与表 22相当。表 23的检出限总体水平比表 22差,这是由于空白溶液制备效果下降所致。
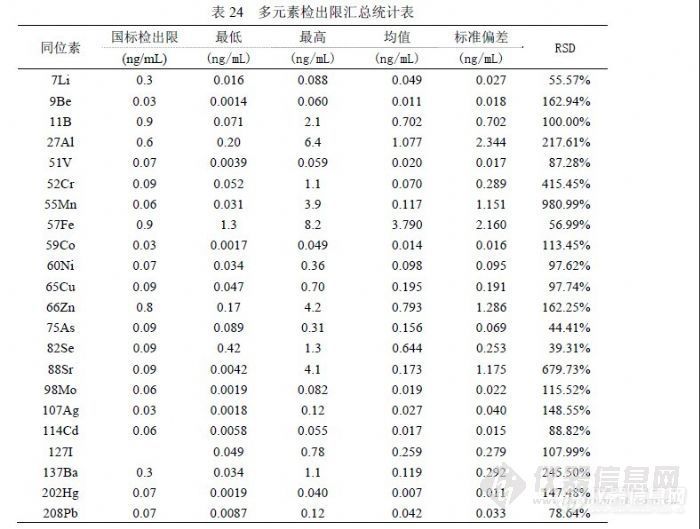
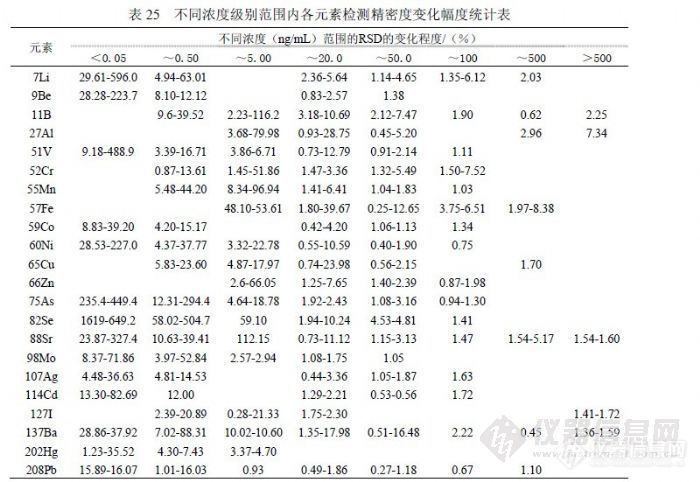
表 22是 2009年 6月实验结果,使用一个空白溶液进行 12次进样统计的检出限参数,正常情况下相当于仪器检出限。与《 GB/T 8538-2008》提出的参数比较,202Hg、137Ba、114Cd、107Ag、98Mo、88Sr、59Co、60Ni、 51V、9Be、7Li等同位素远低于国标,它们在一般矿物土壤中含量很低,且容易清洗不易污染仪器背景,同时这些同位素也具有相对足够高的灵敏度; 52Cr、55Mn、65Cu、66Zn、208Pb等同位素与国标接近,它们的特点是空白信号值较大但 RSD较好,应受纯水、硝酸影响的较大; 11B、27Al、75As的精密度都很好,但由于空白信号值过大导致检出限参数不理想,其原因主要是制备空白溶液的纯水所含杂质含量过高,同时受环境因素影响,可能空气中粉尘进入溶液产生污染;57Fe、82Se是检测同位素中灵敏度比较低的两个,57Fe的空白信号值较大,主要的影响因素是纯水和硝酸,若检测 10ng/mL以下浓度的 Fe需要纯度等级更高的纯水、硝酸及 1000级(或更好)洁净条件,82Se的检测精密度受到 83Kr干扰,改善的方法就是提高氩气纯度,通过验证若不扣除 83Kr的干扰检出限反而会好,因为信号值增加改善了精密度,但能够使低浓度样品分析结果偏高至少一个数量级。有观点认为空气中粉尘影响 ICP-MS的检测结果不可理解,这是因为在 ICP-MS技术应用之前原子吸收所检测浓度相对较高,感受不到环境粉尘的影响,实际上微电子行业的实验室在其制备、检测环境都达到 1000级洁净,更高的达到 100级洁净,在试剂纯度方面采用 SEMI国际标准按 C1~C12级,最差的 C1级相当于优级纯。其有关资料认为影响检测结果的沾污一般有四类:颗粒(来自设备、超净间空气、工艺气体和化学试剂、去离子水),金属(来自设备、超净高纯试剂、离子注入、灰化、反应离子刻蚀),有机物(来自超净间气体、光刻胶残渣、贮存容器、工艺化学试剂),自然氧化物(来自超净间湿度、去离子水冲洗)。我们在实验中也观察到,暴露空气的空白溶液随放置时间增加,元素 Pb、Ba、Cd、Zn、Cu、Ni、B、Cr等不同程度随之浓度升高,另外内标也含有检测元素,并且不同时期配制的内标溶液含有干扰物的浓度不同。经常出现的情况是,某些元素的空白信号值中有很大部分来自在线内标溶液,如 27Al有 70%、11B有 70%、51V有 40%、55Mn有 50%、57Fe有 50%、59Co有 50%、60Ni有 80%、65Cu有 70%、66Zn有 90%、82Sr有 50%、127I有 60%、137Ba有 50%等。表 22的仪器条件处于灵敏度较高时期,其内标信号可达 30万,后出于延长检测器寿命考虑取下屏蔽圈,灵敏度降为 30%。表 23是 12月以 12份独立空白溶液检测后统计的检出限参数,每份溶液均 1次进样连续采集 3次取均值参加统计。与表 22相比,能够确定 6月实验的空白溶液中 Li、B、As、Ag受到污染,因为表 23的空白信号偏低幅度与灵敏度下降幅度不符,而且这 4个同位素尽管精密度不及表 22,但获得了更好的检出限参数。同位素 137Ba、114Cd、98Mo、88Sr、66Zn、65Cu、 60Ni、59Co、57Fe、55Mn、52Cr、51V等都因精密度变差而获得比表 22更差的检出限参数,27Al的原因类似,其空白信号虽有效下降但没有抵消精密度变差的效果。9Be因灵敏度下降而导致检出限上升,208Pb和 202Hg的检出限水平与表 22相当。表 23的检出限总体水平比表 22差,这是由于空白溶液制备效果下降所致。![]() 表24汇总统计了多次实验合计的11个检出限实验数据,统计出最高值、最低值和均值用以和《 GB/T 8538-2008》提出的参数比较,寻找某些元素实验结果不佳的原因。同位素 57Fe、75As和 88Sr以我们实验室目前纯水制备条件无法做到国标提出的参数,75As和 88Sr使用两种纯水机的二次水直接上机做连续 12次采样,以当前仪器的灵敏度和精密度条件刚好与之相当,而 57Fe即使如此也达不到 0.9ng/mL的水平,虽然如此并不影响这 3个同位素的日常检测,因为样品浓度通常高出《 GB/T 8538-2008》提出的检出限参数至少一个数量级以上。同位素 82Se比较奇怪,在扣除 83Kr的干扰后信号值会非常小,精密度也非常差,这是检出限参数无法与国标接近的主要原因,但《EPA200.8》提出的 Se最低检出限是 1.3ng/mL,同《GB/T 8538-2008》相差两个数量级,实验数据也表明 0.09 ng/mL确实难以达到。注意到《 GB/T 8538-2008》中检测 Se选用 77Se,在丰度方面其 7.5%对应 82Se的 8.84%并不占优势,在干扰方面 82Se仅受到 83Kr的干扰,而 77Se分别受到 77ArCl(24.5%)和 14N+63Cu(68.8%)的两方面干扰,在 ICP-MS中因为使用 Ar2和 HNO3,加上水中一般 Cl-含量会很高,所以干扰会非常强,这也是包括 EPA在内很多文献都采用 82Se的主要原因,像《GB/T 8538-2008》这样使用 77Se还能够获得 0.09ng/mL的检出限参数,是非常有意思和有必要讨论的问题。同位素 11B、27Al、55Mn、60Ni、65Cu、66Zn、137Ba和 208Pb也受到纯水背景一定程度的影响,还受到硝酸纯度的影响,其检出限变化的主要原因是环境洁净程度的影响,包括容器、环境空气、附近设备等因素,溶液暴露空气的方式和时间与检测效果相关。表 24的汇总结果代表了实验室基本检测条件和日常操作水平,其统计的均值应做为方法检出限参数,检测限统计应在检出限基础上乘 3.3系数。 6.4 精密度我们分别从标准溶液和样品的角度对不同浓度的检测精密度进行统计,结果见表 25和表 26。
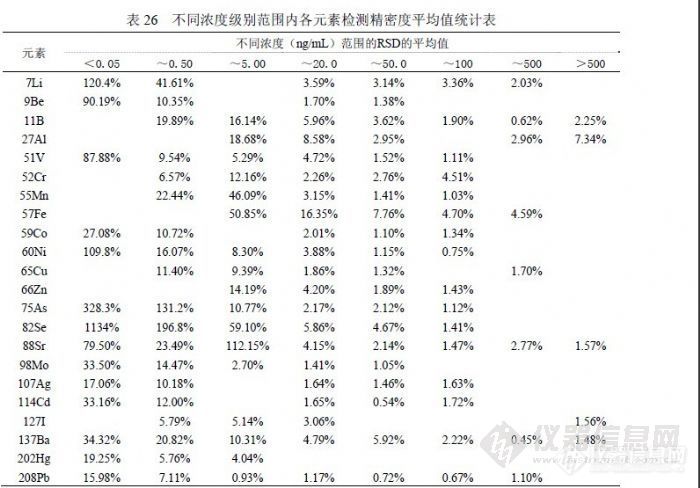
表24汇总统计了多次实验合计的11个检出限实验数据,统计出最高值、最低值和均值用以和《 GB/T 8538-2008》提出的参数比较,寻找某些元素实验结果不佳的原因。同位素 57Fe、75As和 88Sr以我们实验室目前纯水制备条件无法做到国标提出的参数,75As和 88Sr使用两种纯水机的二次水直接上机做连续 12次采样,以当前仪器的灵敏度和精密度条件刚好与之相当,而 57Fe即使如此也达不到 0.9ng/mL的水平,虽然如此并不影响这 3个同位素的日常检测,因为样品浓度通常高出《 GB/T 8538-2008》提出的检出限参数至少一个数量级以上。同位素 82Se比较奇怪,在扣除 83Kr的干扰后信号值会非常小,精密度也非常差,这是检出限参数无法与国标接近的主要原因,但《EPA200.8》提出的 Se最低检出限是 1.3ng/mL,同《GB/T 8538-2008》相差两个数量级,实验数据也表明 0.09 ng/mL确实难以达到。注意到《 GB/T 8538-2008》中检测 Se选用 77Se,在丰度方面其 7.5%对应 82Se的 8.84%并不占优势,在干扰方面 82Se仅受到 83Kr的干扰,而 77Se分别受到 77ArCl(24.5%)和 14N+63Cu(68.8%)的两方面干扰,在 ICP-MS中因为使用 Ar2和 HNO3,加上水中一般 Cl-含量会很高,所以干扰会非常强,这也是包括 EPA在内很多文献都采用 82Se的主要原因,像《GB/T 8538-2008》这样使用 77Se还能够获得 0.09ng/mL的检出限参数,是非常有意思和有必要讨论的问题。同位素 11B、27Al、55Mn、60Ni、65Cu、66Zn、137Ba和 208Pb也受到纯水背景一定程度的影响,还受到硝酸纯度的影响,其检出限变化的主要原因是环境洁净程度的影响,包括容器、环境空气、附近设备等因素,溶液暴露空气的方式和时间与检测效果相关。表 24的汇总结果代表了实验室基本检测条件和日常操作水平,其统计的均值应做为方法检出限参数,检测限统计应在检出限基础上乘 3.3系数。 6.4 精密度我们分别从标准溶液和样品的角度对不同浓度的检测精密度进行统计,结果见表 25和表 26。![]() 对两个实验的标准溶液和样品溶液的检测结果精密度进行统计然后按浓度高低分类统计,观察各元素在不同的浓度范围内精密度变化趋势。浓度由低到高分如下范围(ng/mL):低于 0.05、0.05到低于 0.50、0.50到低于 5.00、5.00到低于 20.00、20.00到低于 50.00、50.00到低于 100.0、100.0到低于 500.0、500.0以上等。表 26的平均值表现了虽浓度上升精密度越练越好的趋势,在浓度低于 0.05 ng/mL时各元素无法控制在 30%以内,浓度在 20.00ng/mL以上各元素精密度都可控制在 30%以内,在(0.50~20.00)ng/mL之间各元素表现不一样。结合表 25的变化幅度情况,精密度较好的元素有 11B、51V、59Co、60Ni、65Cu、75As、88Sr、98Mo、127I、137Ba、208Pb 等,这个范围内统计 9Be、107Ag、114Cd、202Hg没有意义,57Fe、82Se的浓度在 5.00ng/mL以下精密度确定较差(在 30%规定之外),7Li、27Al、 52Cr、55Mn在浓度低于 5.00ng/mL情况下存在较多不确定因素,高于 5.00ng/mL时精密度容易获得控制。
对两个实验的标准溶液和样品溶液的检测结果精密度进行统计然后按浓度高低分类统计,观察各元素在不同的浓度范围内精密度变化趋势。浓度由低到高分如下范围(ng/mL):低于 0.05、0.05到低于 0.50、0.50到低于 5.00、5.00到低于 20.00、20.00到低于 50.00、50.00到低于 100.0、100.0到低于 500.0、500.0以上等。表 26的平均值表现了虽浓度上升精密度越练越好的趋势,在浓度低于 0.05 ng/mL时各元素无法控制在 30%以内,浓度在 20.00ng/mL以上各元素精密度都可控制在 30%以内,在(0.50~20.00)ng/mL之间各元素表现不一样。结合表 25的变化幅度情况,精密度较好的元素有 11B、51V、59Co、60Ni、65Cu、75As、88Sr、98Mo、127I、137Ba、208Pb 等,这个范围内统计 9Be、107Ag、114Cd、202Hg没有意义,57Fe、82Se的浓度在 5.00ng/mL以下精密度确定较差(在 30%规定之外),7Li、27Al、 52Cr、55Mn在浓度低于 5.00ng/mL情况下存在较多不确定因素,高于 5.00ng/mL时精密度容易获得控制。![]() 综合上述情况,检测方法的精密度参数与检测浓度相关,使用较高浓度做实验当然会获得较好的精密度参数,但对低浓度样品则毫无意义,在样品的检测浓度变化范围较大的情况下,求证单一的精密度参数同样没有任何意义。所以研究方法的精密度参数以表 26的统计结果为评估依据。 6.5 干扰校正《EPA200.8》在干扰校正方面做如下表述“特征质谱干扰也要进行校正。不管有没有加入盐酸,所有样品都要进行氯化物干扰校正,因为环境样品中氯化物离子是常见组分。”并为此提出 75As、111Cd、52Cr、Pb、98Mo、82Se、 51V、115In的校正方法。在我们检测的元素中,通过实验研究得出与《EPA200.8》不完全一致的结果,硝酸介质条件下检测 51V如果按 EPA200.8方法进行校正,样品检测信号会出现较大幅度增加,增加幅度与标准溶液不匹配,统计数据中出现了 100ng/mL的标准溶液信号增加 3%时 2ng/mL的样品溶液信号可能增加 1500%。这是因为 51V和 52Cr的灵敏度相当,但样品中 52Cr信号值远高于 51V,干扰了校正过程,导致出现偏差。通过实验观察,使用硝酸介质进行水分析,根据饮用水背景特点,51V、52Cr、98Mo、 Pb通常情况下应不予校正,如果使用盐酸或王水介质因存在大量的 Cl将导致由 Cl引起的干扰过强必须进行干扰校正,75As、82Se、114Cd可以根据同位素丰度、实验经验数据制定干扰校正方程,在使用内标校正前先行校正。具体方法如下。 75As=75M-3.066×[77M-0.826×(82M-1.001×83M)]; 82Se=82M-1.001×83M; 114Cd=114M-0.08587×117M;其中,“M”是元素通用符号。75As主要受到 77ArCl的干扰,而 77ArCl又受到 82Se的干扰,82Se还因氩气纯度因素受到 83Kr干扰;114Cd主要受到 Sn干扰,因为 Sn在 114也由贡献,可以通过检测 120Sn、118Sn、117Sn确定 114Sn的信号值然后扣除,我们选择检测 117Sn。测试过程中另外一个重要干扰是“基体效应”干扰,主要是含盐量过高、某种组分浓度过大等因素使目标物信号受到抑制,由于各目标物和内标等相互间抑制程度不一致,使目标物检测结果出现偏离。消除基体效应的方法有两种,一种是基体匹配,想办法使标准溶液的基体与样品相同或相近,在同一条件下检测达到校正目的,过程比较复杂;另一种是稀释样品,消除原有基体的影响,操作简单,但如果目标物含量较低,有可能稀释后因灵敏度不足导致检出困难。 7 总结通过实验研究明确了以下几个主要问题,其一使用 ICP-MS技术可以检测饮用水中 Fe元素,获得主要方法参数:检出限 6.89ng/mL、检测限 13.77 ng/mL、精密度(3.37~10.25)%、加标回收率(96.29~103.41)%、线性范围( 0~2000)ng/mL;其二使用 ICP-MS技术可以检测饮用水中 Hg元素,浓度在 0.25 ng/mL的样品应使用不同浓度范围的标准曲线检测,样品浓度在
综合上述情况,检测方法的精密度参数与检测浓度相关,使用较高浓度做实验当然会获得较好的精密度参数,但对低浓度样品则毫无意义,在样品的检测浓度变化范围较大的情况下,求证单一的精密度参数同样没有任何意义。所以研究方法的精密度参数以表 26的统计结果为评估依据。 6.5 干扰校正《EPA200.8》在干扰校正方面做如下表述“特征质谱干扰也要进行校正。不管有没有加入盐酸,所有样品都要进行氯化物干扰校正,因为环境样品中氯化物离子是常见组分。”并为此提出 75As、111Cd、52Cr、Pb、98Mo、82Se、 51V、115In的校正方法。在我们检测的元素中,通过实验研究得出与《EPA200.8》不完全一致的结果,硝酸介质条件下检测 51V如果按 EPA200.8方法进行校正,样品检测信号会出现较大幅度增加,增加幅度与标准溶液不匹配,统计数据中出现了 100ng/mL的标准溶液信号增加 3%时 2ng/mL的样品溶液信号可能增加 1500%。这是因为 51V和 52Cr的灵敏度相当,但样品中 52Cr信号值远高于 51V,干扰了校正过程,导致出现偏差。通过实验观察,使用硝酸介质进行水分析,根据饮用水背景特点,51V、52Cr、98Mo、 Pb通常情况下应不予校正,如果使用盐酸或王水介质因存在大量的 Cl将导致由 Cl引起的干扰过强必须进行干扰校正,75As、82Se、114Cd可以根据同位素丰度、实验经验数据制定干扰校正方程,在使用内标校正前先行校正。具体方法如下。 75As=75M-3.066×[77M-0.826×(82M-1.001×83M)]; 82Se=82M-1.001×83M; 114Cd=114M-0.08587×117M;其中,“M”是元素通用符号。75As主要受到 77ArCl的干扰,而 77ArCl又受到 82Se的干扰,82Se还因氩气纯度因素受到 83Kr干扰;114Cd主要受到 Sn干扰,因为 Sn在 114也由贡献,可以通过检测 120Sn、118Sn、117Sn确定 114Sn的信号值然后扣除,我们选择检测 117Sn。测试过程中另外一个重要干扰是“基体效应”干扰,主要是含盐量过高、某种组分浓度过大等因素使目标物信号受到抑制,由于各目标物和内标等相互间抑制程度不一致,使目标物检测结果出现偏离。消除基体效应的方法有两种,一种是基体匹配,想办法使标准溶液的基体与样品相同或相近,在同一条件下检测达到校正目的,过程比较复杂;另一种是稀释样品,消除原有基体的影响,操作简单,但如果目标物含量较低,有可能稀释后因灵敏度不足导致检出困难。 7 总结通过实验研究明确了以下几个主要问题,其一使用 ICP-MS技术可以检测饮用水中 Fe元素,获得主要方法参数:检出限 6.89ng/mL、检测限 13.77 ng/mL、精密度(3.37~10.25)%、加标回收率(96.29~103.41)%、线性范围( 0~2000)ng/mL;其二使用 ICP-MS技术可以检测饮用水中 Hg元素,浓度在 0.25 ng/mL的样品应使用不同浓度范围的标准曲线检测,样品浓度在 - 05ng/mL以下其质量控制性能不适用 30%的 RSD,其常规主要方法参数:检出限( 0.003~0.007)ng/mL、检测限( 0.007~0.03)ng/mL、精密度( 5.20~
- 43)%、加标回收率(70.26~115.92)%、线性范围(0~0.30)ng/mL;其三目前水分析能够定量检测的微量元素有(检出限、检测限单位均ng/mL): 7Li(0.049,0.17)、9Be(0.011,0.037)、27Al(1.1,3.6)、51V(0.020,0.067)、 52Cr(0.070,0.24)、55Mn(0.12,0.40)、57Fe(3.8,13)、59Co(0.014,0.047)、 60Ni(0.098,0.33)、65Cu(0.20,0.66)、66Zn(0.80,2.7)、75As(0.16,0.53)、 82Se(0.65,2.2)、88Sr(0.18,0.58)、98Mo(0.019,0.64)、107Ag(0.027,
- 114Cd(0.017,0.057)、127I(0.26,0.87)、137Ba(0.12,0.40)、202Hg(0.0070,0.024)和 208Pb(0.042,0.15)等,样品浓度在 20.0ng/mL以上检测精密度可以控制在 30%以内。通过方法研究确定了半定量检测和定量检测的方法,确定了检测原始记录必须的格式和内容,规范运用这些方法能够满
足实验室水分析检测上述 20项微量元素的需要。 ICP-MS仪器经过多年发展,在性能上有了很大进步,产生了碰撞池、预四极杆、冷焰、可变分辨率等技术的方便运用,灵敏度、精密度也获得较大提高,极大地拓展了检测范围,出于提高水分析金属元素检测的水平和效率,降低检测成本,应持续开展 ICP-MS检测方法的研究。就像水分析微量元素检测一样,在原 18基础上增加 Fe、Hg所增加的成本远低于原方法(分别使用原子吸收和原子荧光)所有的成本,而检测效率极大提高,只是 ICP-MS检测的难度有所增加,对 ICP-MS检测提出更高的技术要求。主要参考文献
[1] K E 贾维斯,A L 格雷,R S 霍克,等著,电感耦合等离子体质谱手册[M].尹明,李冰 译.北京:原子能出版社,1997,5-15.
[2] EPA Method 200.8. Determination of trace elements in waters and wastes by Inductively coupled plasma-mass spectrometry. Revision 5.4. EMMC Version. 1994.
[3] 张岚等.GB/T 8538-2008 饮用天然矿泉水检验方法.北京:中国标准出版社,2009年 3月.
[4] 尹明等.DZ/T 0130.6-2006 地质矿产实验室测试质量管理规范 第 6部分:水样分析.北京:中国标准出版社,2006年8月.
[5]黄可明,李东雷等.水分析中 ICP-MS在线内标对分析结果的影响[J]. 生命科学仪器,2008,6(11):49~51.
[6] ThermoFisher等编,ICP-MS培训讲义[M].