
获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
分析人生第一次:重读一份十年前的工作进展报告
齐云龙
(中国科学院研究生院生命科学学院 北京100049)
十年有多久?
人生又有几个十年?
十年之前你在何方,
十年之后你又将去向何处?
我们都很难忘记自己的第一份工作,那时候我们满怀热情,充满了好奇和想象,很多时候缺乏经验,或许出现过很多失误,对于偶尔得到的一些进展,我们感到由衷的欣慰——也许很多时候,在很多年之后回头再看的时候,感觉当初自己做的工作是何其的简单……或许最初我们不知道我们付出的努力能否有什么象样的成果;或许很多年后才实然醒悟:正是因为当初付出的某些努力,才会成就我们的今天……
十年之前,我大学刚刚毕业,当时学的是生物化工专业,对电化学还不甚了了……第一次做的分析工作是采用伏安极谱法测定代号为“ECD”和“EC”的放射性原料物质(现在,我当然知道,这三个字母在分析中其实更广泛的含义是电化学测定(Electrochemical detection))。
当时电化学的方法在企业中应该似乎还不够广泛,当时采用的是那种滴汞电极,由于其对环境存在一定的影响而在应用上受到了限制,所用的伏安极谱仪则是天津的一位教授为了推广该方法而让我们免费试用的。所以,我们当时对自己的工作缺少经验,对于能否采用该仪器对产品进行测定没有一点自信。而四个月后,检测较为稳定时,因其简单易用,较为可靠,指导我们的一位北师大的老师力主把该方法推荐为药典检测新标准……
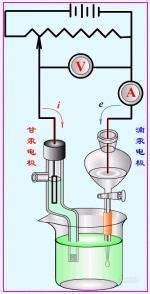 |  |
伏安极谱仪的原理图 | 从网上找的图片,2002年我们用的伏安极谱仪与上图相似,是天津一位大学教授为了推广让我们试用的…… |
| 左图为我们现在用的上海辰华的电化学分析仪相近的图片,与十年前已经不可同日而语,但是,前者的研究和应用却为后者的发展打下了基础! |
更为重要的是,因为那四个月的经验,在九年之后我又有机会进入中科院学习电化学相关的新的知识,我惊讶地发现,原来我们十年前做的当时看来微不足道的事情竟然还可能深深影响到十年后的生活……
下面,是当时的一份总结报告,很难得还能从当时保存的纸质文档里找了出来(电子版当时保留在公司电脑未能做私人备份),我把这份文档用扫描仪重新扫描识别,保留下来作为纪念!
如今重读起来,仍旧有很多东西值得思考!虽然当时不懂得去查阅相关的文献来学习相关知识,更远远不具备科研的思维,但是,因为关注到的那些细节,我得到了更理想的结果!!重读起来,还感叹自己当时所想到的那诸多方面……
每一个进步都需要一定的基础,我从中感受最深的就是,不要轻视任何一份工作,任何的分析工作都需要认真、严谨的态度,小至刷试剂瓶这样的工作以及周围的通风、温度、湿度等状况都可能会对检测的结果产生极为重要的影响……在分析实验当中应该关注每一步细节——我们会因为注意到这些细节而学到更多东西……
附后:写于十年前的一份总结报告:
ECD伏安极谱仪法测定进展报告
我正式触ECD的伏安极谱法测定是从(2002年——作者注)8月8日开始的,至今已经有约两个月,从对此几乎一无所知开始,现在已经初步了解了这一方法来测定ECD含量的测定过程,以下是我对方法的一些认识:
1. ECD波峰位置的确认
因为原来已经进行过一段时间的实验,我原认为实验已经比较成熟。受原来测定质量标准的误导,一开始认为ECD波峰应出现的位置约在-0.6 V左右(原来使用的极谱仪为示波极谱仪),但实际上经反复确认(采用添加原料使对应波峰增高的方法)最终确认为-0.2 V左右。
2.溶剂的变化
由于在测定中ECD前出现两个波峰(之后经进一步实验,我认为前一波峰也有可能是氧波导致,因为用盐酸溶解也会除ECD之外的波峰,这一步因为是刚开始做,有一些草率,之后也未进行详细对比实验:水解程度如何?何时水解效果就开始很显著),初步认为是由ECD发生水解所致,同时也为与ECD中亚锡含量的测定相一致,我们试采用盐酸来进行溶解。
3.溶剂浓度的影响
在盐酸的浓度上我们实验了1N、0.5N、0.1N等,之后一直采用1N的盐酸来溶解,以避免其发生水解。
4. Kit中其它成分的影响
因为极谱法测定出现的波形较复杂,药盒中的成分较多,我们采用混合添加药盒成分的方法来验证底液成分(主要是亚锡)对测定的影响,结论是其它成分对测定无明显影响(由此也可以推断以上述方法也可以测定EC等的含量)。
5.仪器准确度的确认
考虑到ECD本身不太稳定,我们拟采用一种较稳定的物质来进行验证,因为对极谱法本身不太了解,我们根据《药物检验方法》中极谱法可以用来测定硫代硫酸钠,于是我们采用硫代硫酸钠来验证仪器的稳定性,结果显示:在较低浓度下有一定误差,但在较高浓度下结果很合理(详见实验数据),实验证明:仪器本身是没有太大问题,只是应该考虑试样本身的性质和浓度线性范围等因素。
6.氮气保护的作用
排除了对仪器的怀疑之后,我们首先用它来测定亚锡的含量,经查阅相关资料,结合亚锡本身易被氧化的性质,我建议|涂配制用的盐酸要用氮气饱和外,在测定时也用氮气轻吹,以建立一个较为稳定的环境(既要保证惰性环境义要使被测定溶液液面没有大的波动)。实验证明上述方法较为有效,测定结果十分理想,不仅回归曲线线性关系好,测定结果也十分理想!这一步不仅进一步验证了仪器的可信度,同时也给了我们很大的信心,让我们一度陷入困境的实验又重新拥有了进行的动力!可以算作实验进程中一个比较可喜的突破!
但是经过实验,我们也验证了氮气对ECD的测定影响的作用尚不太明显。
这次实验一个重要收获是,让我们进一步认识到了环境因素(如氧气)的影响。
7.档位选择对测定结果的影响
如果样品本身比较稳定,则无论选择分辨率高还是低的档位对结果都不会造成太大的影响,但如果样品线性关系不太好,又考虑“原点电流”影响的话,则越高的档位显示的线性关系会越差,理论上测定对结果应该没有太大的影响,但实际测定高低档位却有所差别!而且有时候差别很大。所以一般测定时可选择多个档位平行测定;在不超过最大电流值时可以选择较高档位对比电流值变化情况,而低档位则会出现较好的线性关系,可以用来作实验结果确认!
8.同步测定的重要性
对照品和药盒的配制和测定应该尽可能同步,否则对结果可能会产生较大的影响,例如我们曾作过对比,同一样品25 μg/mL时,前后相隔一小时测定,则结果一次较准,另一次则高达32μg/mL时,换算为药盒ECD含量分别为1.0mg/瓶和1.28 mg/瓶,可见结果差别很大。尤其当所选择的档位对电流较敏感,而且配制的浓度又比较低时,换算成药盒含量时结果相差更大!
9. ECD溶解后曲线关系的验证
这一步实验对以后的影响至关重要,尤其是朱老师建议的用多个浓度同时测定来对比的一次实验,从根本上验证了ECD溶解之后从低浓度到高浓度明显呈现一条上弯的曲线,经第二天测定与第一天对比,个人认为发生的理化作用较为明显,经过近一周的跟踪对比实验,结果显示曲线关系直线化越来越明显,而且高浓度电流值会上升,而低浓度电流值则会下降,变化比较不规则。之后我们开始注意时间(相对“同步”测定来讲是一段很长的时间)对测定结果的影响。现在我无法确定一周之后溶液内部究竟发生了什么理化反应,但确实经放置之后会使测定结果合理化。
之后一步是对配制完的样品浓度进行改变,来找一段线性关系比较明显的区间来寻求配制完当时测定的方法(这一步的测定思路也是非常有价值的),经过这一步的实验我们可以得出结论:低浓度下相对原来的浓度呈现的线性关系较快,但是测定时由于要进行换算所以最终结果也有可能会因此产生较大的偏差。所以这也是一种比较冒险的方法,最好不要列入质量标准中。
10.加热与不加热对溶液测定的影响
本实验只作了一组,我认为不太理想,由于配制的样品较多,前后时间相差太久,对对比结果影响太大,以至二者可比性不大,但经加热之后的样品其波峰明显复杂化,出现其它的波峰,(例如我们推测为EC的波峰)较多,且一开始时加热与不加热没有太大区别,而之后则开始出现较大差别,考虑本方法本身实行起来比较复杂,所以放弃了该方法。可以考虑进一步的实验,例如用来测定其它产品时同步进行。
11.叠波的影响
两种物质半波电位小于0.2 V时,两个极谱波就会重叠,不易分辨,我认为EC和ECD就与这种情况相似,但更为复杂,因为ECD和EC在溶液中可能会相互转化! ECD经过加热或用水作为溶剂时就会出现这种情况,由于ECD水解会产生EC,两者电位单独出现时分别为-0.16 V和-0.21 V,但同时出现时则两个波峰合二为一,波峰电极电位大概出现在-0.18 V,而添加EC或ECD时该波峰电流值则都会有所增加,所以在加热或产生水解时结果不会准确!(在ECD测定中ECD波峰前有一个波峰电极电位-0.15 V左右,我们一直认为是氧波,但因为对结果不造成太大的影响,至今没有完全确认,我认为也有可能是ECD的某种水解产物。
12.关于质量标准修改革案
关于质量标准草案的具体修改,我建议尽量简化实验说明部分而采用计算机所给出的数据和图表报告等。因为计算机本身的计算已经很有说服力!只需用数据证明: ECD本身溶解后呈现线性关系,另外证明单标准测定的准确性即可。
13.仪器测量环境的影响
影响极谱法测定的因素很多,包括毛细管管径、汞柱高度、环境温度、氧波、叠波、前波、氢波、极谱极大现象等。由于环境因素对测定的影响很重要,所以实验过程中的变化,如环境温度、毛细管更换、家柱高度、氮气保护与否等都会对结果产生比较明显的影响!应尽量保证实验状况的稳定。
14.数据处理方式的改进
仪器本身的处理方式有一些缺陷,例如缺少“标准品”的数据,打印比较浪费纸张,对比不太清晰,曲线与数据不在同一页面等。经过改进,我修改程序来进行数据的处理,效果较好,同时也有了比较大的选择余地。
15.应注意的问题
实验中有一些制约因素应当注意,实验中曾经出现过的问题有:
a.玻璃管
因为玻璃管较细,虹吸作用很强,容易发生堵塞,温度低时更易发生!
而更换玻璃管之后测定环境则会随之发生变化,所以平时应尽量避免虹吸(放下滴汞瓶时,玻璃管下端不应浸入溶液中),并备有相同规格均一性好的玻璃管。
b.用水
一般用生产部一次水即可,但实验过程中曾出现由于停水而造成实验无法进行的情况,所以实验之前应备有充足的用水。
c.盐酸
确定好应用何种浓度的盐酸后,每次实验前应事先配制好!
d.氮气
由于液氮瓶是封密的,一般不易查觉到所剩余的氮气量。应该注意每次实验之前先要准备好将盐酸用氮气饱和。
e.小烧杯
每次用之前一定要洗干净,以保证不影响测定结果,一般测定应该按从低浓度到高浓度的顺序测定。
f.电极的固定
不能接触杯底的汞滴(以防止反应难以进行);另外铂电极不能接触上面的金属架(以防止电极短路),否则都会导致波形异常;
g.汞的散落和挥蒸发
汞蒸气是剧毒的,实验中应当谨防汞的散落和蒸发,用后的汞要及时回收,实验室要注意通风并可用燃烧碘的方法来进行消毒。
小结: ECD药盒成分含量测定中亚锡的测定进展还算顺利,但ECD含量测定进展却十分缓慢,除其本身性质比较复杂外,与我们对它的认识程度不够以及自己相关专业知识的欠缺有着重要的关系,所以今后对于要测定的物质首先要对其物理、化学等性质进行充分的了解,同时应该加强专业知识的学习,掌握相关检测方法、处理数据的方法以及计算机相关程序的熟练操作,以避免走很多弯路。同时进行实验之前都要进行比较详细、周密的思考和设计,对相关仪器设备作好充分的准备工作,尤其是头脑必须要保持高度的清晰,对每一步要进行的实验有充分的考虑,不打没有准备的仗!这样才能保证实验的效率和最终效果的理想。
近期实验,为以后的测定打下了比较坚实的的基础,但存在的问题也很多,有待进一步认真努力的实验来验证。
(完)
(注:以上总结报告文字完成于2002年10月,即恰是十年之前)












































