获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往
原文由 littalnala(littalnala) 发表:原文由 huangza(huangza) 发表:
不错的话题,单从前处理的角度看,样品消解最终应该为澄清透明的液体;如果从整个检测过程上,应该加一个质控样对比或者加标回收试验验证一下。
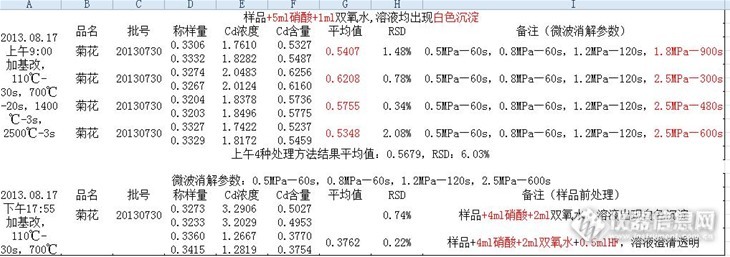
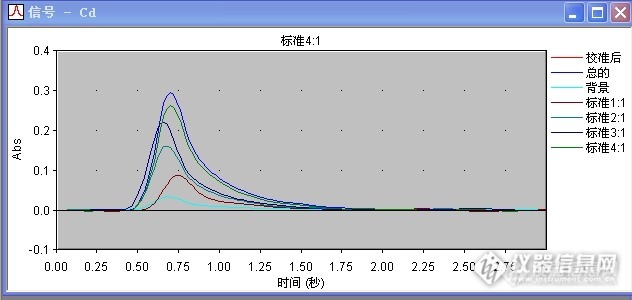
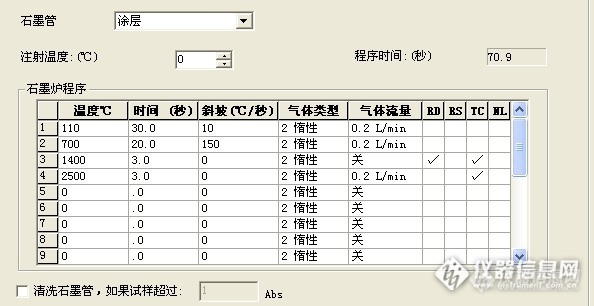
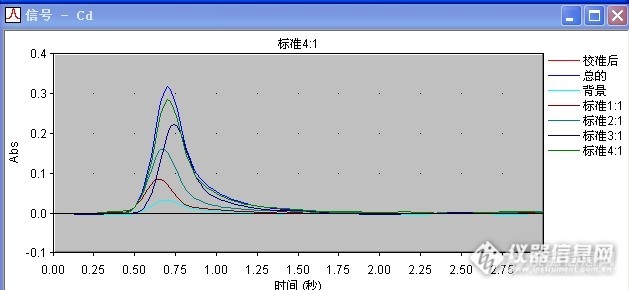
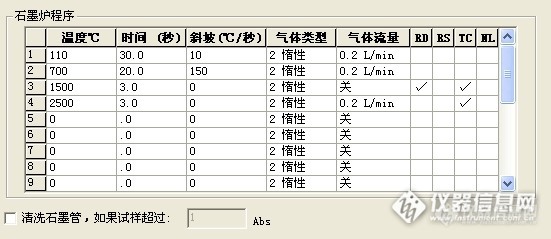
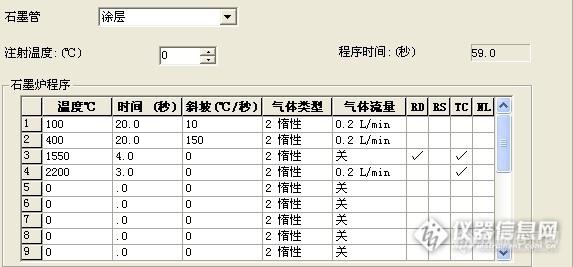
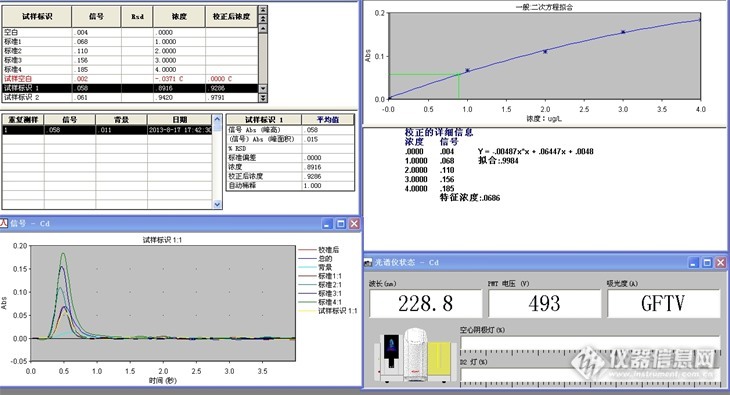
请问下 你做过 用同个样品溶液 上机的时候 采用加基改与不加基改的方法 然后对比两种方法的结果吗?我这边就出现了 加基改,菊花样品的背景峰挺高的,信号一般在0.180左右,不加基改,背景峰反而没那么高,只有0.020左右,虽然都有用塞曼校正,我在想是否是加入的基改与菊花里面的某些成分反应而导致的?
原文由 huangza(huangza) 发表:原文由 littalnala(littalnala) 发表:原文由 huangza(huangza) 发表:
不错的话题,单从前处理的角度看,样品消解最终应该为澄清透明的液体;如果从整个检测过程上,应该加一个质控样对比或者加标回收试验验证一下。
请问下 你做过 用同个样品溶液 上机的时候 采用加基改与不加基改的方法 然后对比两种方法的结果吗?我这边就出现了 加基改,菊花样品的背景峰挺高的,信号一般在0.180左右,不加基改,背景峰反而没那么高,只有0.020左右,虽然都有用塞曼校正,我在想是否是加入的基改与菊花里面的某些成分反应而导致的?
做过加基改与不加基改的情况,我们正好相反。不加基改有些样品的背景值很高,无法计算样品真实值。加基改后,样品背景值相当低,一般还不到0.01,我们用的是硝酸钯做基改,另外我们仪器是氘灯扣背景。
建议你尝试其他基改试试。