维权声明:本文为v3246897原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
话说为了应对市场需求,需要进行硫酸粘杆菌素的检测方法的开发,查找标准方法,大致浏览下用
液质进行检测,基本资源有满足,开始工作。
![]()
1.化合物性质及结构
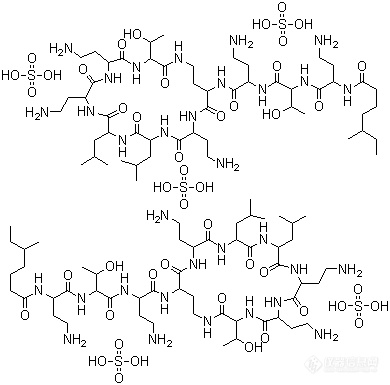
先介绍下硫酸粘杆菌素,分子结构图如下:
![]()
分子式:2(C
52H
98N
16O
13).5(H
2SO
4),分子量:2801.27,纯度80.1%,PH3-7.5范围内稳定,为硫酸粘杆菌素A和硫酸粘杆菌素B的混合物。
2. 配制标样。
用十万分之一的天平,称取硫酸粘杆菌素10.20mg至10mL容量瓶中,用换算成硫酸粘杆菌素浓度C=2=1347.98mg/L,用体积比V
1+V
2=1+4的0.1甲酸乙腈溶液溶解,并配置成10mg/L工作液。
3. 仪器方法建立。
3.1质谱条件
3.1.1寻找母离子
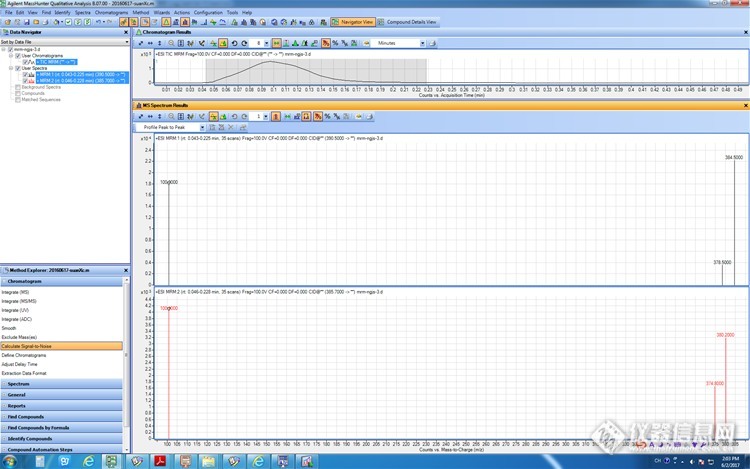
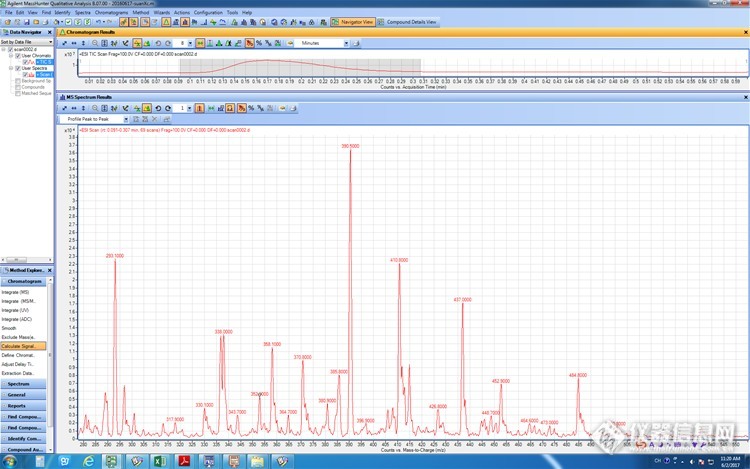
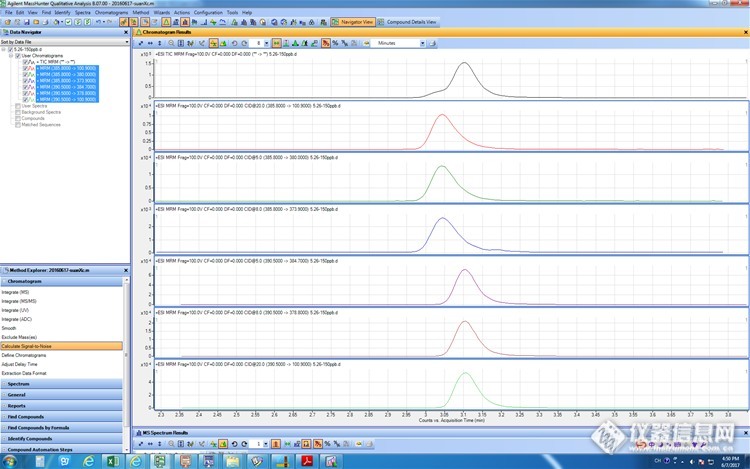
硫酸粘杆菌素是由氨基酸缩合而成的碱性多肽类抗生素,在一级质谱中易与多个H +结合成多电荷正离子,初步选定流动相A1+B2=3+7,接双通,流速0.2mL/min,进浓度为5.0mg/L的标准品1μL,初始Fragment为100,用MS2 SCAN正模式扫描,得到ESI正离子模式全扫描质谱图如图所示,硫酸粘杆菌素的三电荷离子
3 +响应最高,,进而确定它们的母离子(m /z) 分别为390. 5、385. 8。
![]()
3.1.2优化Fragment电压
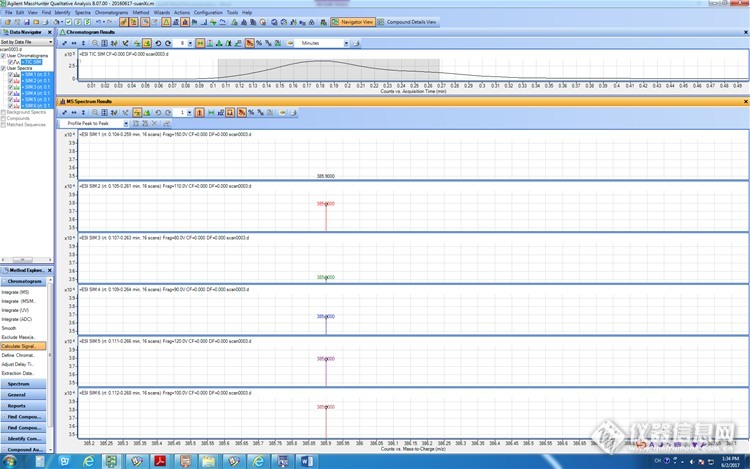
将扫描模式该为MS2 SIM模式,将得到的Precursor Ion输入,驻留时间Dwell设为20,Fragment从1v-200V范围设置,在其他仪器条件不变的情况下进样,得到不同Fragment下的响应强度图,初步选定其Fragment在100V左右,进一步在100左右细化Fragment,最终选定硫酸粘杆菌素A和B的Fragment为110V。
![]()
![]()
3.1.3 ProductIon和CE优化
将扫描模式选为Product Ion模式,将390.5和385.8作为Precursor Ion,MS2 From 100,MS2 To 400(包含母离子),Fragment选定优化好的110V,碰撞能按照CE=左右寻找,得到子离子,选定包含国标的离子,并得到大致CE范围,将扫描模式选为MRM模式,在得到的初步CE左右,进一步优化得到最佳的CE值。
![]()
综上,得到硫酸粘杆菌素的质谱条件,如下表:
表 1 目标化合物的质谱分析条件| No. | 中文名称 | 英文名称 | CAS | 前体 | 产物 | 去簇电压 | 碰撞能 | 加速电压 |
| 离子 | 离子 | (V) | (V) |
| 1 | 硫酸粘杆菌素A | Ceftriaxone Sodium | 1264-72-8 | 390.5 | 384.7* | 110 | 5 | 4 |
| 378.8 | 110 | 8 | 4 |
| 100.9 | 110 | 20 | 4 |
| 2 | 硫酸粘杆菌素B | Ceftriaxone Sodium | 1264-72-8 | 385.8 | 380.0* | 110 | 5 | 4 |
| 373.9 | 110 | 5 | 4 |
| 100.9 | 110 | 20 | 4 |
注:* 为定量离子。3.2 流动相条件
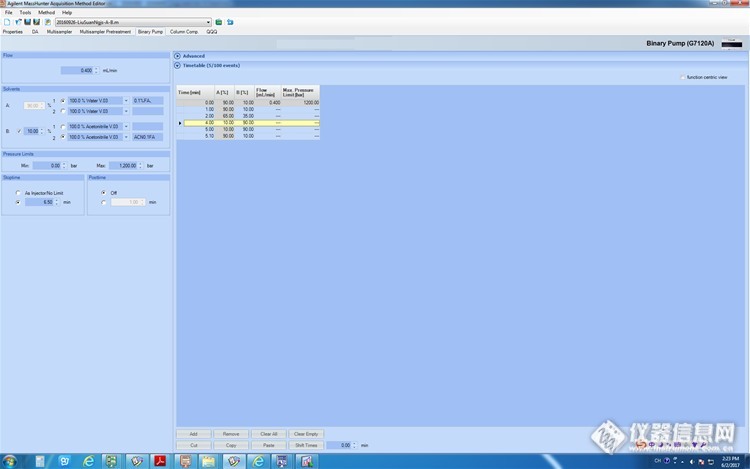
液相条件的寻找,首先确定流动相,在双通的用乙腈+0.1%甲酸水=7+3、乙腈+0.1%甲酸水=5+5、乙腈+0.1%甲酸水=3+7 3种比例条件下看峰型和响应的大小,选定乙腈+0.1%甲酸水=7+3时出峰最好,然后乙腈+0.1%甲酸水=7+3、乙腈+0.1%甲酸水5mmol/L乙酸铵=7+3、0.1%甲酸乙腈+0.1%甲酸水=7+3,出峰强度和峰型与乙腈+0.1%甲酸水=7+3基本一样,确定硫酸粘杆菌素在流动相比例乙腈+0.1%甲酸水=7+3时出峰最佳,先接色谱柱,笔者试验Agilent EclipsePlus C18 RRHD 1.8μm2.1*100mm,Waters HSS T3 1.8μm,Agilent XDB-C18 1.8μm,最终发现Agilent XDB-C18 1.8μm,对目标化合物分离效果及峰型最好。![]()
![]()
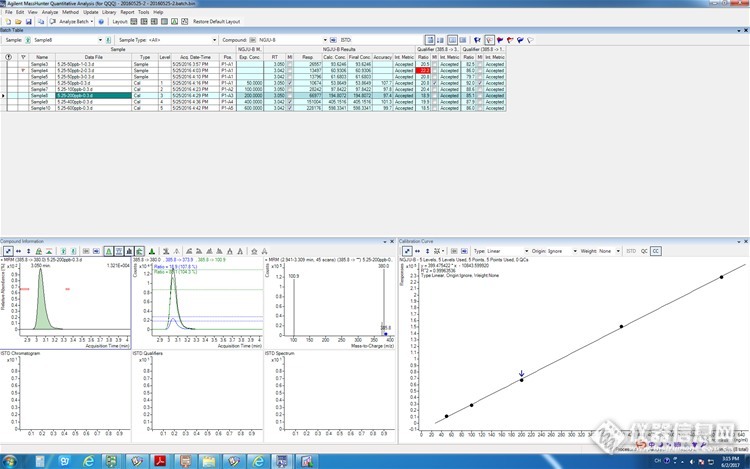
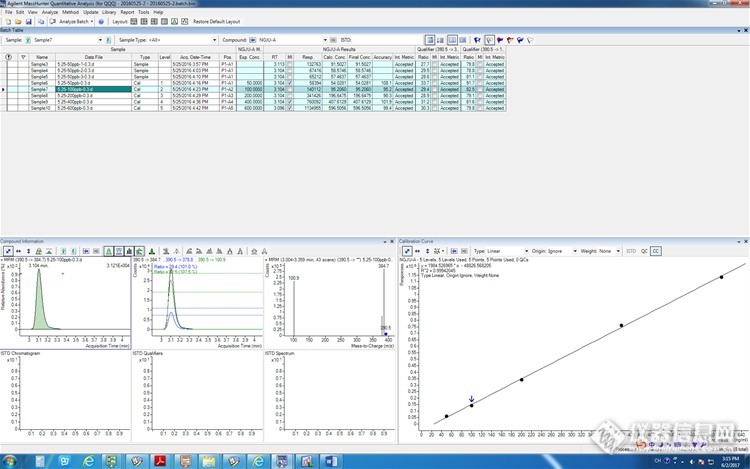
4.用建立好的仪器方法配置标曲
硫酸粘杆菌素 St50μg/L, 100μg/L,200μg/L,400μg/L,600μg/L.
![]()
![]()
4.前处理条件
4.1.化合物分析
粘杆菌素为多肽类抗生素,含有多个氨基,极性很强,易溶于水,微溶于甲醇等有机溶液,PH3-7.5范围内稳定,可用HLB和阳离子交换柱进行净化。4.2试验方法4.2.1 前期试验 通过国标方法及文献资料,选择4%三氯乙酸+4%乙酸铅, 10%三氯乙酸:乙腈=3:7溶液作为提取溶剂, 提取离心后 ,用正己烷净化 ,过500mgHLB ,3mL水淋洗,3mL5%甲醇/水淋洗,6mL甲醇洗脱 35℃旋转蒸干或者N2吹干上机分析,加标小瓶中理论值100μg/L,全程试剂空白,猪肉未知样品,及加标回收,均一样大,sp100μg/L有明显的基质干扰,单纯的标准品35℃旋转蒸干或者N2吹干上机分析,无回收。
4.2.2前期试验分析与总结:
a.最终进样不能吹干或旋干,选择N2(35℃以下)吹至1mL以下;
b.HLB洗脱接受后,溶液较浑浊,说明HLB净化效果不佳;
c.提取液4%三氯乙酸+4%乙酸铅较其他两种浑浊;
d.分析化合物有多个氨基可选择离子交换柱, WCX柱。
4.2.2 再次试验称样5.0g
--- 添加标准品1mg/L×100μL---加入10%三氯乙酸:乙腈=3:7 15mL
----充分混匀,超声20min,离心
----上清液倒入新离心管---- 残渣继续用10%三氯乙酸:乙腈=3:7 10mL提取一遍----合并上清液,用氨水调PH=7
---- 加入10mL乙腈饱和的正己烷,充分混匀,离心
----弃去正己烷层,下层过WCX(200mg,6cc),甲醇水平衡 ----5mL水淋洗
----5mL甲醇淋洗
----6mL甲酸:甲醇=1:4洗脱,接收
----用N2在40℃下吹至1mL左右,用水定容至2mL,过0.22μm滤膜。
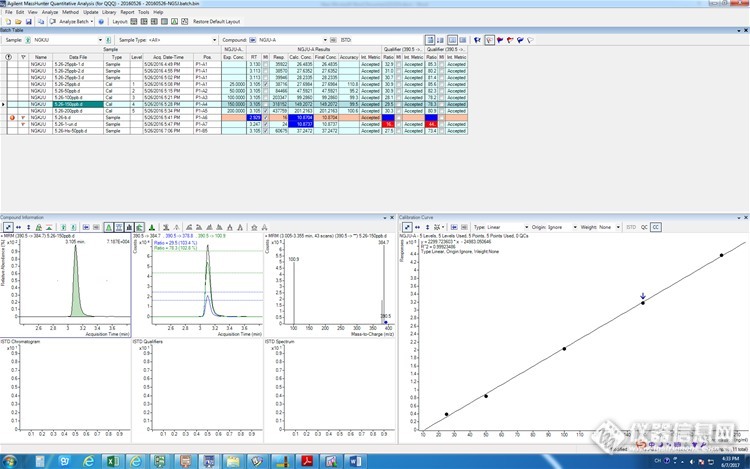
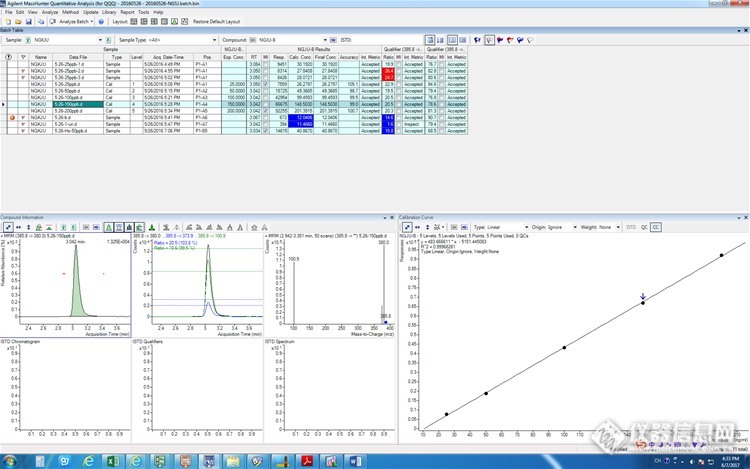
线性范围:25μg/L,50μg/L,100μg/L,150μg/L,200μg/L基质标曲。得到如下谱图:
![]()
![]()
回收率在70%左右。
综上:对于硫酸粘杆菌素,提取试剂用酸性,选用离子交换柱比HLB柱净化干净,定容之前溶液勿吹干(多次失败得出)。
以上,是本人做硫酸粘杆菌素仪器方法开发时的一些经验和体会,希望能给大家带来帮助,也希望大家能多提提意见,共同进步,谢谢大家。![]()