气相色谱应用中,柱流失由于直接影响分离和检测效果,因此一直倍受使用者的关注。本文将就柱流失的原理、流失的评价以及故障盘查等进行剖析和讨论,希望能对大家有所帮助。
什么是柱流失?
日常应用中,我们观察到的流失实际上是系统内污染物或流失物到达检测器的所有信号的总和, 它包含了以下来源:载气、进样口、色谱柱和检测器。那么什么是纯粹的色谱柱流失呢?严格来讲,柱流失是
气相柱固定相趋于热力学稳定的降解反应或热分解反应。大多数
气相色谱柱的固定相由高分子聚合物组成,趋于热力学稳定,长链聚合物的末端具有缓慢的卷曲→成环→脱落的降解反应,这些脱落的固定相碎片即是色谱柱的流失。 2. 流失程度与哪些因素有关?
柱流失程度与固定相种类和色谱柱规格有关,基本规律是:在其它因素都相同的情况下,固定相的极性越高,固定相膜越厚,色谱柱内径越粗,柱长越长,柱流失则越高。就色谱柱规格而言,即固定相的量越高、柱流失则相对较高。此外,柱流失还与柱温以及载气中氧气的浓度有关。高温下,固定相的降解会加快,因此,高温时的流失会比低温时高。氧气是许多固定相降解的催化剂,特别是在高温时段,我们强调载气的纯度和色谱柱安装的气密性, 一方面就是出于氧气会加剧柱流失的考虑
3. 如何评价柱流失?
柱流失情况通常可通过绘制流失曲线来观察和评价。流失曲线是运行程序升温空针所采集的基线,正常情况下,它应当是一条平滑的与程序升温相吻合的曲线,即基线随着柱温升高而提升,当柱温恒定后,基线也稳定在一个水平。
4. 什么检测器适合检查柱流失?
由于对固定相流失碎片的选择性好、灵敏度高,FID和MS检测器可以说是最适合检测固定相的流失的检测器。在色谱柱生产领域,生产厂家普遍采用FID进行出厂前的流失检查,这是因为FID易于操作并且经济耐用,它对柱流失的响应完全可以同MSD媲美。MSD尽管也很灵敏,但影响其输出因素过多(如离子源的清洁度、载气的纯度和流量、质谱参数的设置等等),相比之下,使用FID更容易获得重现和可靠的结果
5. 柱流失高故障盘查
前面提到,真正柱流失的来源只应该是色谱柱,而我们所观察到的流失实际上是到达检测器的所有信号的总和,如果我们发现色谱柱在老化之后流失高,甚至伴随有杂峰和基线波动等现象,这时还不能简单地把问题归结为色谱柱,下面介绍一些故障盘查方法。
高温时流失高
步骤1)重新安装色谱柱。套好柱螺帽和密封垫圈后,重新切割色谱柱;注意不要将手上的污渍油渍带到色谱柱,因为这很可能导致鬼峰。
步骤2)将色谱柱安装到进样口,通好载气,在室温下吹扫约15分钟。对于分流不分流进样口,最好选择分流模式,分流流量可设置为80mL/min,并关闭载气节省(gas saver)功能。如果使用了保护柱,需要检查保护柱与分析柱连接处的气密性,可使用电子检漏计或滴加异丙醇的方法检漏。
步骤3)运行空针程序升温,如果基线在到达最高温度20分钟后回落至可接受的水平,说明载气中氧含量没有异常。如果高温时基线始终不下降甚至一直攀升,应立即降温,进行载气纯度检查和气路检漏。
流失高并伴随鬼峰与杂峰
色谱柱固定相的流失是连续的,一般不会产生一个一个的“流失峰”。绝大部分情况下,我们所观察到的杂峰或鬼峰是来自于色谱柱之外的污染,故障盘查可参考以下方法:
步骤1)将进样口温度降至室温,设置炉温为较低温度如50C,恒温约20分钟,观察基线;将进样口温度提高到高温,如250C,再在50C恒温观察20分钟。如果杂峰信号增加,说明有来自进样口或载气的污染。可重点检查进样口隔垫和衬管,必要时刚换新的。样品瓶隔垫有时也可引入聚硅氧烷类物质的污染,可检查样品瓶里是否有隔垫的碎屑,或比较使用不带隔垫的样品瓶进行排查。
步骤2)检查检测器是否污染。具体步骤是:断开并取下色谱柱,用堵头封住检测器入口。打开检测器,如果继续出现杂峰,则应该立即考虑清洗检测器,并检查检测器所用气体管线的清洁度。
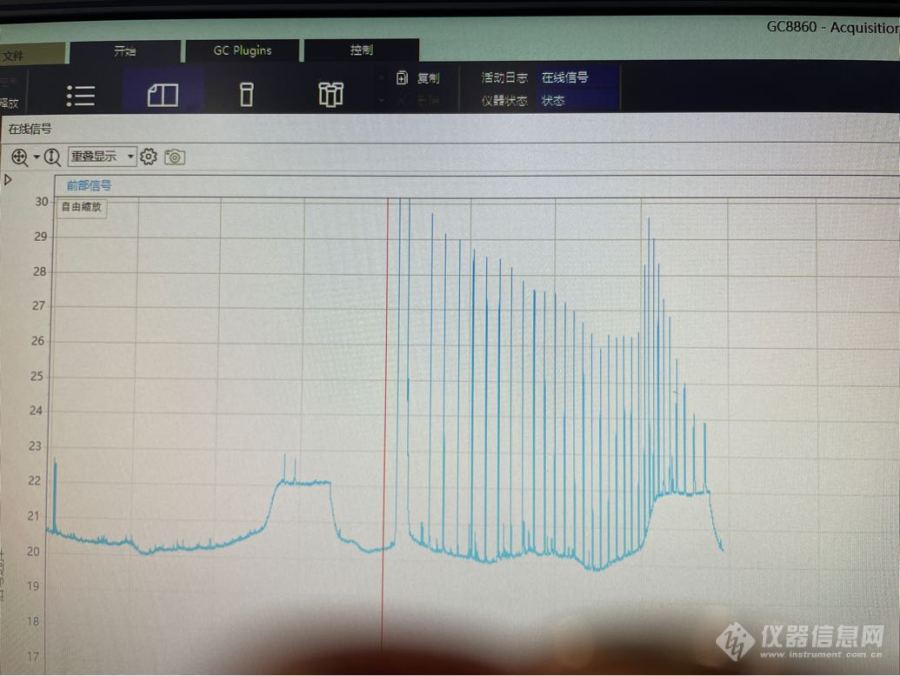
以下是一个来自隔垫流失的实例:
流失高并伴随基线波动
较高的流失并且基线波动通常是色谱柱被污染的征兆,样品中的挥发性差的组分可能不在第一次程序升温流出,而在后面的温度循环中缓慢流出,从而导致较高的波动的基线。遇到这种情况,建议维护和更换进样口衬管与隔垫,色谱柱进样口端切割掉0.5~1米,重新安装并在高温老化色谱2~3小时,观察基线是否恢复正常。
兰线:色谱柱没有经过任何处理的流失曲线
红线:色谱柱前端割掉一圈,高温老化2小时的流失曲线
绿线:再次割掉色谱柱前端一圈,并高温老化2小时的流失曲线