兴奋剂类分析方法系列讲座(20)生物检材中苯丙胺类兴奋剂的$$lc-MS/MS分析 生物检材中苯丙胺类兴奋剂和氯胺酮的
$$lc-MS/MS分析
摘 要 目的:建立生物检材中苯丙胺类兴奋剂和氯胺酮的
液相色谱-串联质谱(
$$lc - MS/MS)分析方法。
方法:生物检材包括血液、尿液和毛发,采用稀释法和液-液提取的前处理方法,应用两个不同的
液相柱,优化
$$lc – MS/MS分析方法,并考察了血液和尿液基质的离子抑制作用。
结果:同时分析苯丙胺和MDA,
液相1在3min内完成,
液相2可用于确认分析或复杂基质分离。尿液稀释法检材用量少,前处理简便快速。毛发中苯丙胺类兴奋剂和氯胺酮的最低检测限(LOD)为0.005~0.05ng/mg。对送检案例检材产妇头发和胎毛进行苯丙胺类兴奋剂和氯胺酮的分析。
结论:本方法可用于生物检材中苯丙胺类兴奋剂和氯胺酮的同时分析,血、尿等生物检材的离子抑制作用是影响本方法灵敏度的主要原因。
本文由司法部司法鉴定科学技术研究所的向平、卓先义、沈保华、马栋和沈敏与复旦大学上海医学院的姜宴等研究人员共同完成,是上海市自然科学基金项目的部分内容,文中的生物检材除血、尿还做了多项人体和胎儿毛发中含量的测定值得参考。现全文介绍如下。
前 言
苯丙胺类兴奋剂是一类低分子量、合成的苯丙胺衍生物的统称,包括苯丙胺( amphetamine )、甲基苯丙胺(methamphetamine )、MDA(3,4-methylenedioxyamphetamine)和MDMA(3,4-methylenedioxymethamphetamine ),具有兴奋中枢神经和周围神经、抑制食欲、致幻等作用。苯丙胺类兴奋剂在舞厅等娱乐场所滥用非常严重,并存在与氯胺酮 (ketamine)混合使用。目前涉及苯丙胺类兴奋剂和氯胺酮的死亡案件不断上升,急需建立快速、简便、灵敏、准确地同时分析生物检材中的苯丙胺类兴奋剂和氯胺酮的方法。
液相色谱-串联质谱(
$$lc-MS/MS )是
液相分离结合串联质谱检测的新型分析仪器,可直接分析难挥发性化合物、极性化合物、热不稳定化合物和大分子化合物,分析范围广,且不需衍生化步骤[1~3]。目前
$$lc-MS/MS在我国较少用于毒物分析。本文研究建立
$$lc-MS/MS同时定性定量分析生物检材(血液、尿液和毛发)中苯丙胺类兴奋剂和氯胺酮的方法。
实 验
1. 材料与方法
1.1材料
氯胺酮、去甲氯胺酮、内标甲基苯丙胺-d5和氯胺酮-d4 ( Cerilliant公司),甲基苯丙胺、苯丙胺、MDMA、MDA和麻黄碱等 (国家麻醉品实验室),乙腈、甲酸和乙酸胺 ( Fluka公司)。API 4000三重四极串联质谱仪
$$lc-MS/MS (Applied Biosystems公司)。
1.2
$$lc-MS/MS方法
1.2.1
液相1
液相柱为Allure Ultra IBD ( Resteck公司) 50mm×2.1 mm × 5 μm,此前接phenomenex的保护柱。流动相为乙腈,缓冲液:20mM乙酸胺:0.1 %甲酸(80:20),恒流200μ1/min 。
1.2.2
液相2
液相柱为Allure PFP Propyl ( Resteck公司) 100mm×2..1 mm× 5μm,此前接phenomenex的保护柱。流动相为乙睛,缓冲液:20mM乙酸胺:0. 1%甲酸 (70: 30 ),恒流200μ1/min。
1.2.3 质谱
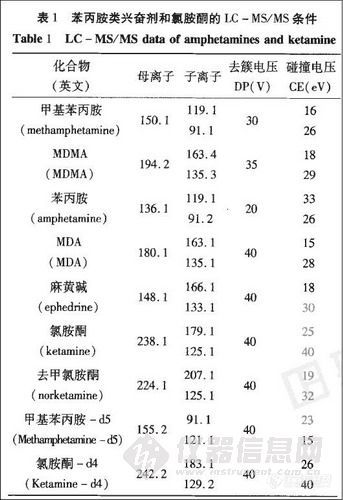
采用电喷雾电离-正离子模式 (ESI+),操作参数分别为:碰撞气 (Collision Gas ) 5;气帘气 (Curtain Gas ) 25;离子喷射电压 (Ionspray Voltage)4500 V;温度 (Temperature ) 450 ℃。每个化合物选取两个离子对,见表1。
![]()
1.3 样品前处理方法
1.3.1 尿液稀释法
20μl尿液加入1 ml流动相,混旋,离心,取5μ1进样。
1.3.2 血液、尿液提取法
血液或尿液1 ml加入2m1 pH9. 2磷酸缓冲液,加入3. 5ml乙醚,混旋,离心,取有机层,60℃水浴中挥干,加入100μ1流动相,取5μ1进样。
1.3.3 头发提取法
依次用0. 1%十二烷基磺酸钠、o. l%洗洁净、蒸馏水、丙酮洗涤,晾干后剪成约1mm长,备用。空白头发同法处理。头发20mg加0.1 mol HCl 1 ml浸泡过夜,取出后用1 mol NaOH调至中性,加2m1 pH9. 2磷酸缓冲液,以下同1.3.2项处理。
1.4 生物检材基质的离子抑制作用检验
分别用
液相流动相配制成1ng/ml和l0ng/ml的MDMA对照品稀释液,各取100μl×4份加至带衬管的
液相进样瓶中。空白血液和尿液各1 ml × 8份,采用1.3.2步骤提取,挥干后,4份为一个浓度组,残余物中分别加入100 μ1配制的对照品稀释液溶解。各取5μ1进样。
2 结果与讨论
2.1结果与评价
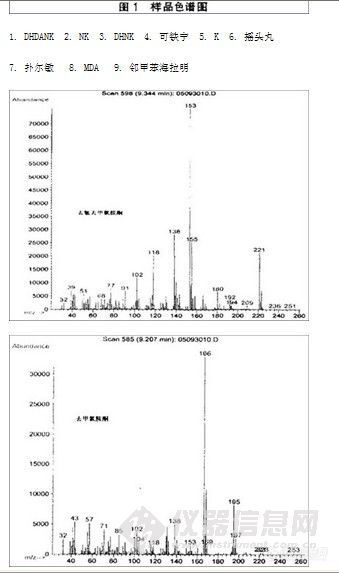
所建立的
$$lc-MS/MS方法分离良好 (图1),
液相1在3 min内完成,与常规的GC/MS方法相比,大大节约了分析时间。
液相2可用于确认分析或复杂基质分离。
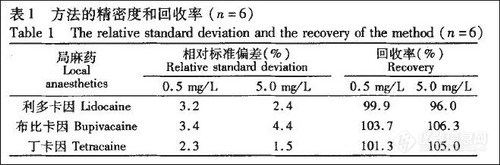
MS/MS可以进行两次离子选择作用,通常称之为多反应监测 (multiple reaction monitoring,MRM),即通过MSl选择一定质量母离子,与气体碰撞断裂后,再经MS2选择一定质量的子离子,这样大大提高了分析的专一性和灵敏度,可以分析复杂基质体系中的待测物。本方法中每个化合物选取两个离子对,采用第一个离子对定量分析,以第二个离子对峰强度信噪比 (S/N) 大于3确定最低检测限(LOD),增加了分析的准确性和特异性。
尿液稀释法检材用量少,前处理简便快速。血液、尿液提取法经:过提取纯化,可减少离子抑制。
本方法可同时分析苯丙胺和MDA,克服了
气质联用仪(GC-MS)方法中由于分子量小、极性强而必须衍生化才能检测的缺点,且灵敏度比GC-MS [4] 高近100倍,同时加入甲基苯丙胺-d5和氯胺酮-d4作内标,定量分析准确 (表2)。
![]()
2. 2 生物检材基质的离子抑制作用
基质的离子抑制作用是
$$lc/MS-MS分析中存在的技术难题,是分析物信号损失主要原因之一,影响方法的灵敏度。参考Hendrickson [5] 的方法,本文以MDMA为目标物,采用血液和尿液为基质,观察基质的离子抑制作用。将对照品溶液进样的峰面积设为A,血液和尿液样品提取后加入相同量的对照品后进样的峰面积设为B,抑制作用(matrix effect, ME ) (%) = B/A×100%。结果见表3。
![]()
由表3可看出,血、尿等生物检材的离子抑制作用是影响本方法灵敏度的主要原因,尿液样品高于血液,这可能是由于尿液中的无机盐离子成分相对较多引起。所以,在进行
$$lc-MS/MS分析时,适当的生物检材前处理过程非常必要。
2. 3案例应用
案例1 某公安分局送检头发样本,经去污处理、水解和提取后,GC-MS分析阴性。将提取残余物中加入流动相,采用
$$lc-MS/MS分析,则检出甲基苯丙胺、氯胺酮和去甲氯胺酮。
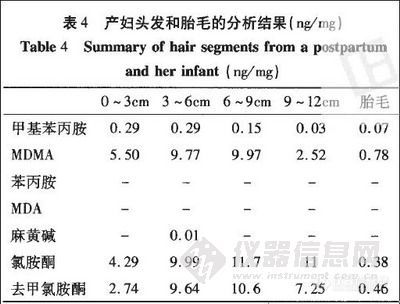
案例2 某妇女在怀孕期间一直滥用“摇头丸”,胎儿产下后有明显的烦躁不安等戒断症状。生产后的第2天紧贴根部剪取产妇头发和胎毛。产妇头发总长约15cm,从根部起按3cm分段分析,结果见表4
![]()
头发的生长速度大约为1~1.2cm/月[6],由此表明,该产妇在怀孕期间一直滥用以MDMA和氯胺酮为主的兴奋剂。孕妇滥用药物不仅会影响胎儿生长,而且对胎儿今后的成长造成严重伤害。有报道从产妇和胎毛中同时检出尼古丁和吗啡类生物碱 [7,8],同样,本案例胎毛中苯丙胺类兴奋剂和氯胺酮的浓度均小于产妇。所分析的头发段中未检出苯丙胺和MDA是因为药物进入毛发的程度与药物的亲脂性密切相关 [9],苯丙胺和MDA的极性强所以很难进人头发。Stefanc Gentili [4] 在分析12份甲基苯丙胺阳性头发样品中,仅有1例检出苯丙胺;57份MDMA阳性头发样品中仅有1例检出MDA。
![]()



































 】
】