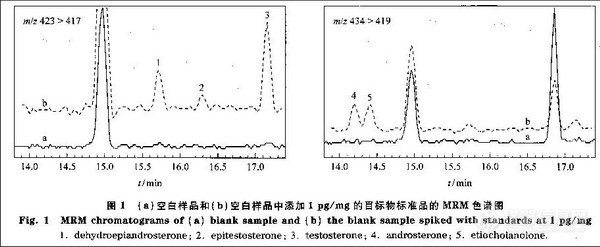
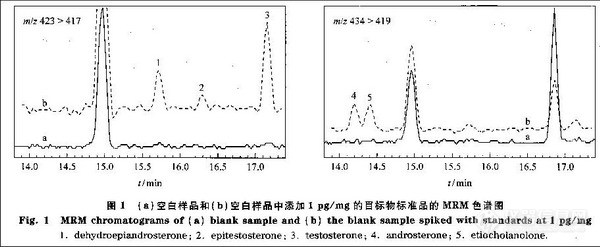
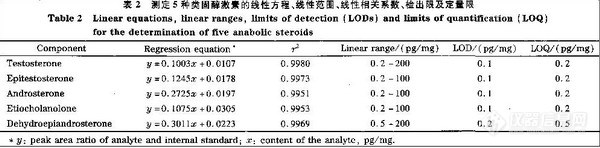
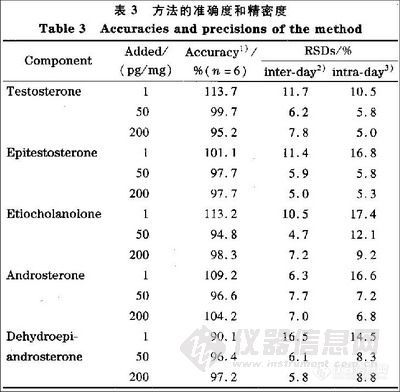
兴奋剂类分析方法系列讲座(26)(下)微波萃取-气相色谱法测定尿液中的苯丙胺类毒品 2. 2. 4 溶剂用量对萃取率的影响
分别取环己烷2, 4, 6, 8, 10, 12 mL 作为萃取溶剂,在40 ℃下萃取10 min, 其余步骤同“1.5.1”节所述。结果表明:随着萃取溶剂用量的增加,萃取率明显增加,大于6 mL 以后,萃取率变化不大。故环己烷用量选择6 mL。
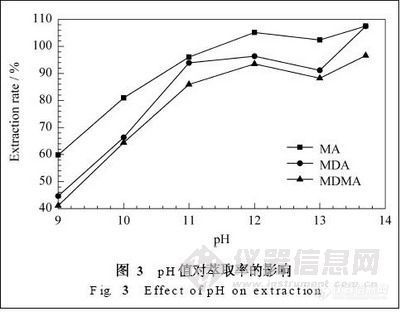
2. 2. 5 尿液pH值对萃取率的影响
取6 mL 环己烷作为萃取溶剂,在40 ℃的萃取温度下保持10 min,分别调节尿液的pH值为9, 10,11, 12,13和13.7,其余步骤同“1.5.1”节所述,结果(见图3)表明:在尿液的pH 值大于12 时, 萃取率变化不大。故选择pH值为12。
![]()
2. 3 标准曲线和最低检出限
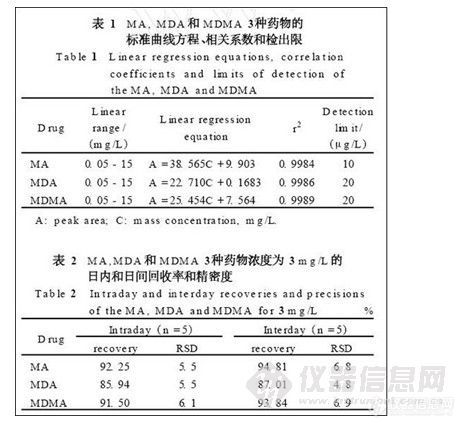
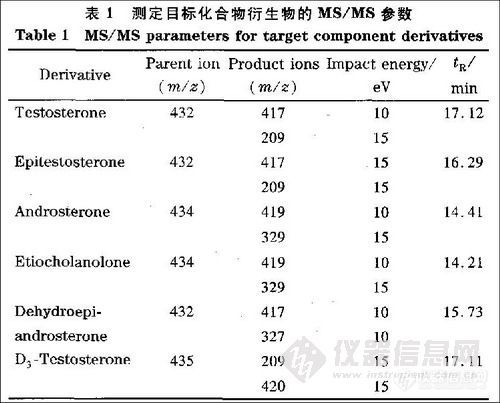
向空白尿液中加入MA, MDA 和MDMA 的混合标准溶液,配制尿液中3种药物质量浓度分别为0.05, 0.1, 0.2, 0.4, 0.5, 1, 2, 3, 4, 5, 10, 15 m g /L 的系列标准溶液。按“1.5.1”节方法处理样品, 然后进行GC-F ID 分析。以药物峰面积(A )对药物质量浓度(C, m g /L )绘制标准曲线, 并求得直线回归方程和相关系数。最低检出限为按“1.5.1”节步骤处理样品所能检测到的尿液中的药物浓度(以3倍信噪比计算) , 结果见表1。3 种药物浓度为3 m g /L的日内和日间回收率和精密度见表2, 其中日内 ( In traday)为1 d 内重复测定5次的回收率和精密度, 日间 ( In te rday)为5 d 内冻融5 次测定的回收率和精密度。
![]()
2. 4 方法的回收率及精密度的测定
取空白尿液,准确加入MA,MDA 和MDMA 的混合标准溶液, 分别配制药物浓度为1, 3, 5 m g /L的尿液各5份,按“1.5”节所述两种方法处理样品,并进行GC-FID 分析,计算平均回收率和精密度,结果见表3。
![]()
2. 5 在法医案例中的应用
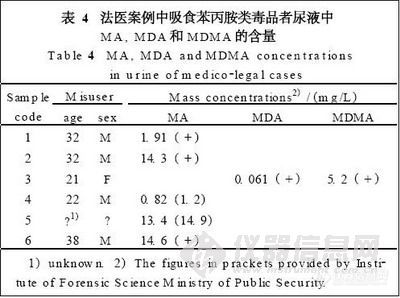
将该方法应用于测定法医案例中6例吸食苯丙胺类毒品者的尿液中MA,MDA 和MDMA 的含量,并且与公安部物证鉴定中心提供的数据(采用GC-MS测定)进行了比较。分析结果见表4 (括弧中的数据为公安部物证鉴定中心提供的数据,“ + ”为阳性结果) ,其中2号样品为冰毒吸食者的尿样,色谱图见图12c; 3号样品为摇头丸吸食者的尿样, 色谱图见图12d。
![]()
2. 6 GC-M S 定性分析
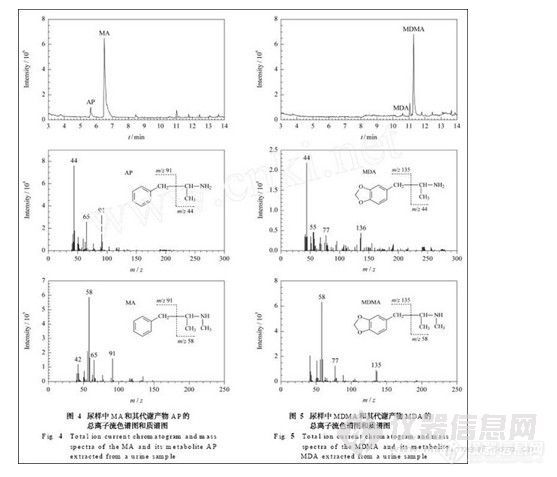
实验过程中,为了进一步增强检验结果的可靠性,我们还同时采用GC-MS 的全扫描模式对微波萃取的吸食苯丙胺类毒品者的尿液样品进行了分析。根据其保留时间和质谱图进一步确定了检材中的毒品成分。图4为表4中2号样品的总离子流色谱图和质谱图(冰毒的主要成分为MA,MA 的代谢产物为苯丙胺(AP ) ) ,图5为表4中3号样品的总离子流色谱图和质谱图(摇头丸的主要成分为MD 2MA,MDMA的代谢产物为MDA ) 。
![]()
3 结论
本文采用微波萃取技术对人体尿液中甲基苯丙胺、3, 42亚甲二氧基苯丙胺和3, 42亚甲二氧基甲基苯丙胺3种常见的苯丙胺类毒品的分析进行了研究,通过对微波萃取溶剂、萃取温度、尿液pH 值等微波萃取条件的优化,建立了同时检测人体尿液中甲基苯丙胺、3, 42亚甲二氧基苯丙胺和3, 42亚甲二氧基甲基苯丙胺的微波萃取2
气相色谱分析方法。该法简单快速、高效,重现性好和回收率高。将该方法应用于6例苯丙胺类毒品滥用者的尿液的检测,
获得了理想结果。
![]()