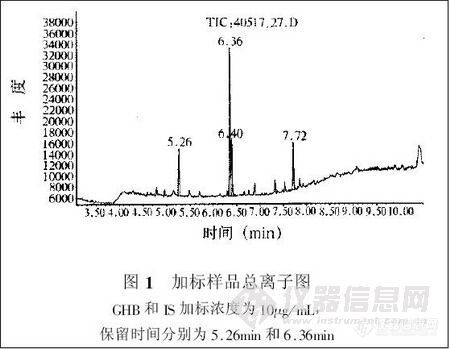
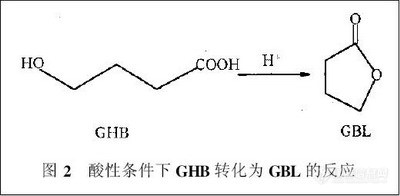
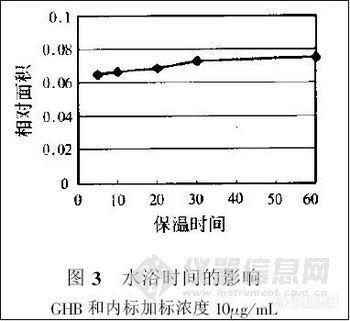
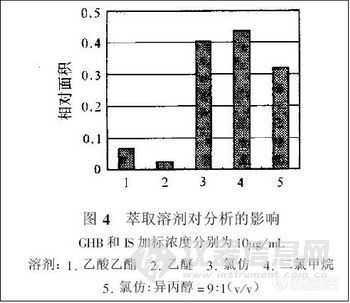
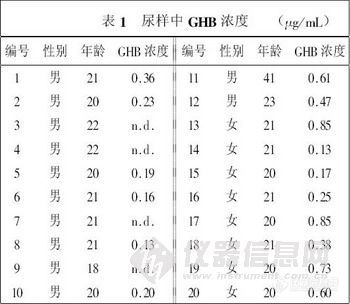
兴奋剂类分析方法系列讲座(38)固相萃取同时提取尿中的39种药毒物 固相萃取同时提取尿中的39种药毒物
摘 要
目的::建立固相萃取方法同时提取尿中的39种药毒物并用高效
液相色谱法进行分析。
方法:以多沙普仑为内标, 1mL尿样经小柱固相萃取, 用HPLC进行分析。
结果:39种药毒物可同时从尿中提出, 内源性物质不干扰测定。其绝对回收率除吗啡外均大于70%;天内及天间精密度均小于10%;检测限1~15ng/mL;线性相关系数在0.9977以上。
结论:本法快速、简便、重现性好, 空白干扰小, 可用于实际案例的药物毒物筛选。
本文由张玉荣1, 梁晨1, 金琦芸2, 郭幼梅2, 张润生1, , 王跨陡1, , 王威1, , 严松茂1
(1、上海市刑事科学技术研究所, 2、上海刃复旦大学药学院药物分析教研室,)共同协作完成。第一作者张玉荣为上海市刑事科学技术研究所副所长,主要从事体内、外药毒物的分离检测和研究的高级工程师。
前 言
固相萃取作为从生物样品中提取净化微量药物或其代谢物的方法, 已被广泛地应用于生物样品的检测中, 其中尤以与HPLC技术结合用于生物样品的分析引人注目〔1~4〕。目前已报道的固相萃取文献大多只涉及到几种或某一类药毒物的分析〔2~4〕, 本文希望通过扩大药物的覆盖面, 建立一种可快速、同时测定多种药毒物的切实可行的并具有通用性的方法, 用同一种前处理方法同时提取尿中的多种药毒物, 以满足实际检案的需要。
实 验
1 材料和方法
1.1 仪器
HP1100系列高效
液相色谱仪(配置G1315光电二极管阵列检测器)。
1.2 对照标准品和试剂
对照品及内标多沙普仑均由中国药品生物制品检定所提供, 39种对照品名称详见表1;取三乙胺100mL加水1200mL混合, 用冰醋酸调节pH8.0, 最后加水至1500mL, 配制成三乙胺缓冲液;固相萃取小柱(HLB 1mL/30g)购自Waters公司。
1.3 方法
1.3.1 样品处理
依次用甲醇、水各1mL通过固相萃取小柱, 吸取1mL尿样上样后依次用1mL 2%氨水5%甲醇的水、1mL5%甲醇的水清洗, 将固相萃取小柱离心甩干(3000r/min ,3min), 最后用1mL二氯甲烷洗脱。洗脱液加人内标储备液3μL在50℃水浴中空气流下吹干, 残渣用50μL甲醇溶解, 取10μL进样。
1.3.2 色谱条件
采用Lichrospher 100 RP-18e(250mm×4mm×5μm)色谱柱;二极管阵列检测器,检测波长为230nm、250nm, 同时进行紫外扫描;柱温50℃ ;流动相A 甲醇:水:三乙胺缓冲液= 50:46:4 , 流速0.8mL/min;而流动相B 甲醇:水:三乙胺缓冲液 = 75:16:9, 流速0.6mL/min。
2 结果
2.1 色谱分离
在选定的HPLC条件下, 药物色谱峰都得到了良好的分离, 流动相A、B下各药毒物的保留时间范围分别为4.99~36.3min和5.43~28.28min。空白尿样经提取后在两个色谱条件下不干扰测定。
2.2 线性关系
以药物峰面积与内标峰面积之比(Y)对样品中药物浓度(X)作图(略), 得39种药毒物的回归方程和相关系数, 各种药物的线性良好, 相关系数在0.9977以上.。
2.3 精密度和回收率
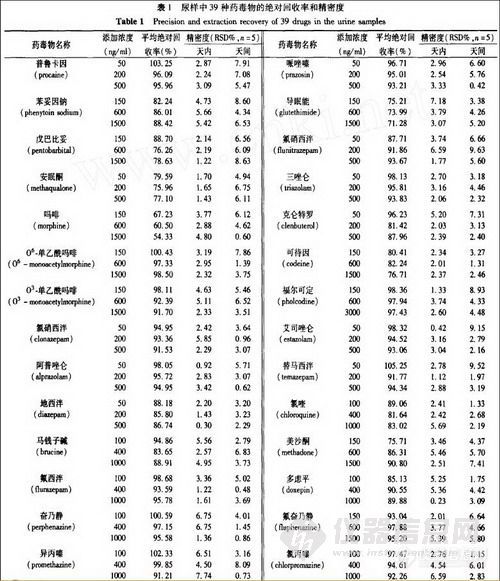
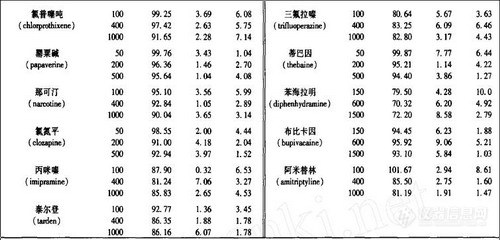
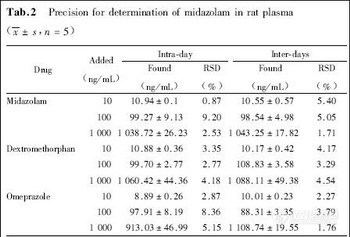
分别加药毒物储备液配成高、中、低三个浓度的尿样, 按1.3.1项样品处理方法操作。每个浓度一天同一次操作中分析5份, 得天内精密度;不同天内各分析1份, 分析5天, 得天间精密度。同时将相应理论浓度的标准溶液进样10μL, 对应的色谱峰与内标峰面积比之比为绝对回收率, 见表1。
![]()
![]()
下列资料由“仪器信息网”负责搜集整理,版权所有。请勿复制粘贴,否则视为侵权
文章类别:
阅读次数:17
文章日期:2009-9-13 12:15:39
固相萃取同时提取尿中的39种药毒物
摘 要
目的::建立固相萃取方法同时提取尿中的39种药毒物并用高效
液相色谱法进行分析。
方法:以多沙普仑为内标, 1mL尿样经小柱固相萃取, 用HPLC进行分析。
结果:39种药毒物可同时从尿中提出, 内源性物质不干扰测定。其绝对回收率除吗啡外均大于70%;天内及天间精密度均小于10%;检测限1~15ng/mL;线性相关系数在0.9977以上。
结论:本法快速、简便、重现性好, 空白干扰小, 可用于实际案例的药物毒物筛选。
本文由张玉荣1, 梁晨1, 金琦芸2, 郭幼梅2, 张润生1, , 王跨陡1, , 王威1, , 严松茂1
(1、上海市刑事科学技术研究所, 2、上海刃复旦大学药学院药物分析教研室,)共同协作完成。第一作者张玉荣为上海市刑事科学技术研究所副所长,主要从事体内、外药毒物的分离检测和研究的高级工程师。
前 言
固相萃取作为从生物样品中提取净化微量药物或其代谢物的方法, 已被广泛地应用于生物样品的检测中, 其中尤以与HPLC技术结合用于生物样品的分析引人注目〔1~4〕。目前已报道的固相萃取文献大多只涉及到几种或某一类药毒物的分析〔2~4〕, 本文希望通过扩大药物的覆盖面, 建立一种可快速、同时测定多种药毒物的切实可行的并具有通用性的方法, 用同一种前处理方法同时提取尿中的多种药毒物, 以满足实际检案的需要。
实 验
1 材料和方法
1.1 仪器
HP1100系列高效
液相色谱仪(配置G1315光电二极管阵列检测器)。
1.2 对照标准品和试剂
对照品及内标多沙普仑均由中国药品生物制品检定所提供, 39种对照品名称详见表1;取三乙胺100mL加水1200mL混合, 用冰醋酸调节pH8.0, 最后加水至1500mL, 配制成三乙胺缓冲液;固相萃取小柱(HLB 1mL/30g)购自Waters公司。
1.3 方法
1.3.1 样品处理
依次用甲醇、水各1mL通过固相萃取小柱, 吸取1mL尿样上样后依次用1mL 2%氨水5%甲醇的水、1mL5%甲醇的水清洗, 将固相萃取小柱离心甩干(3000r/min ,3min), 最后用1mL二氯甲烷洗脱。洗脱液加人内标储备液3μL在50℃水浴中空气流下吹干, 残渣用50μL甲醇溶解, 取10μL进样。
1.3.2 色谱条件
采用Lichrospher 100 RP-18e(250mm×4mm×5μm)色谱柱;二极管阵列检测器,检测波长为230nm、250nm, 同时进行紫外扫描;柱温50℃ ;流动相A 甲醇:水:三乙胺缓冲液= 50:46:4 , 流速0.8mL/min;而流动相B 甲醇:水:三乙胺缓冲液 = 75:16:9, 流速0.6mL/min。
![]()