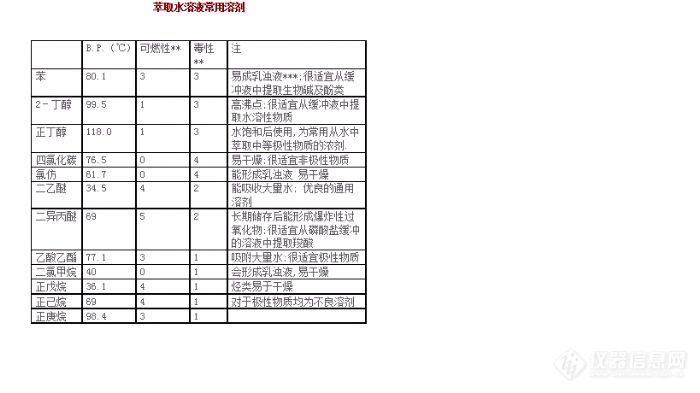
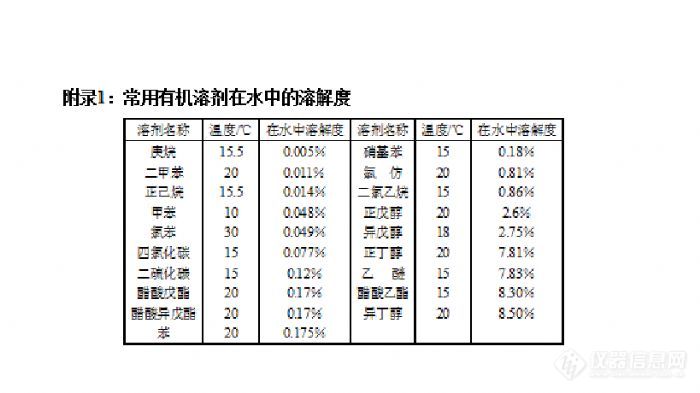
有机实验室常用仪器与使用--旋转蒸发仪
旋转蒸发仪,主要用于在减压条件下连续蒸馏大量易挥发性溶剂。尤其对萃取液的浓缩和色谱分离时的接收液的蒸馏,可以分离和纯化反应产物。旋转蒸发仪的基本原理就是减压蒸馏,也就是在减压情况下,当溶剂蒸馏时,蒸馏烧瓶在连续转动。结构:蒸馏烧瓶可是一个带有标准磨口接口的梨形或圆底烧瓶,通过一高度回流蛇形冷凝管与减压泵相连,回流冷凝管另一开口与带有磨口的接收烧瓶相连,用于接收被蒸发的有机溶剂。在冷凝管与减压泵之间有一三通活塞,当体系与大
气相通时,可以将蒸馏烧瓶,接液烧瓶取下,转移溶剂,当体系与减压泵相通时,则体系应处于减压状态。使用时,应先减压,再开动电动机转动蒸馏烧瓶,结束时,应先停机,再通大气,以防蒸馏烧瓶在转动中脱落。作为蒸馏的热源,常配有相应的恒温水槽。