后续将有专属客服与您沟通!
关注微信公众号查看留言进度 接收留言处理通知
0
ID:silva
行业:其他
积分:0升级还需100积分
声望:0升级还需100声望
注册时间:0000-00-00
最后登录时间:0000-00-00
ID:fengsix
ID:victo
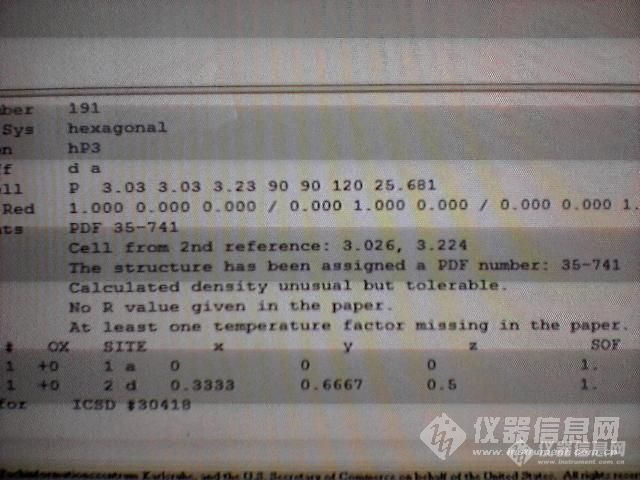
原文由 ustb 发表:原文由 yesky 发表:ustd老兄: 把2d和1a的参数输入到哪个软件里啊谢谢你用的什么软件?Diamond、atom、BS和Crystal studio都不用这个参数,你只要把原子位置输进去就可以了,如果用JEMS就要用到这个参数。请问:以上这些软件哪个好用一些? 直接有很多的不同化合物晶格结构的最好了!就不需要我们费事去建立这些咚咚了。可以回复我的Mail:shengbaoqiu@126.com
原文由 yesky 发表:ustd老兄: 把2d和1a的参数输入到哪个软件里啊谢谢你用的什么软件?Diamond、atom、BS和Crystal studio都不用这个参数,你只要把原子位置输进去就可以了,如果用JEMS就要用到这个参数。请问:以上这些软件哪个好用一些? 直接有很多的不同化合物晶格结构的最好了!就不需要我们费事去建立这些咚咚了。可以回复我的Mail:shengbaoqiu@126.com
你用的什么软件?Diamond、atom、BS和Crystal studio都不用这个参数,你只要把原子位置输进去就可以了,如果用JEMS就要用到这个参数。
ID:guxinfu
ID:usaorange
ID:dilong399
ID:sendtony6



































 直接有很多的不同化合物晶格结构的最好了!就不需要我们费事去建立这些咚咚了。可以回复我的Mail:shengbaoqiu@126.com
直接有很多的不同化合物晶格结构的最好了!就不需要我们费事去建立这些咚咚了。可以回复我的Mail:shengbaoqiu@126.com