获得0积分,您同时完成了每日任务,有额外的积分奖励,请前往APP领取
立即前往

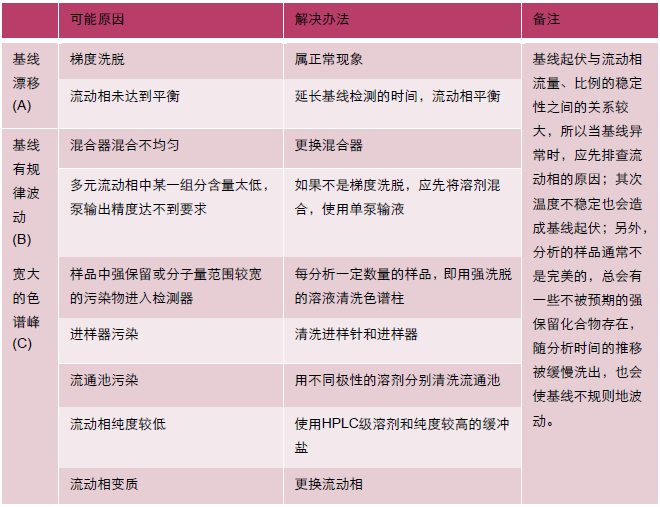
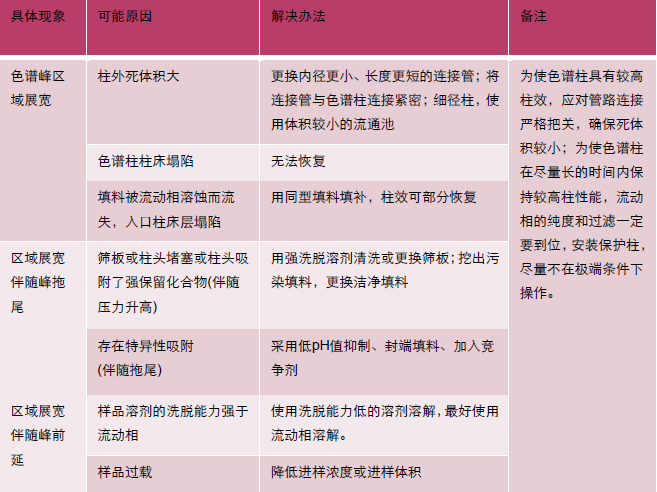
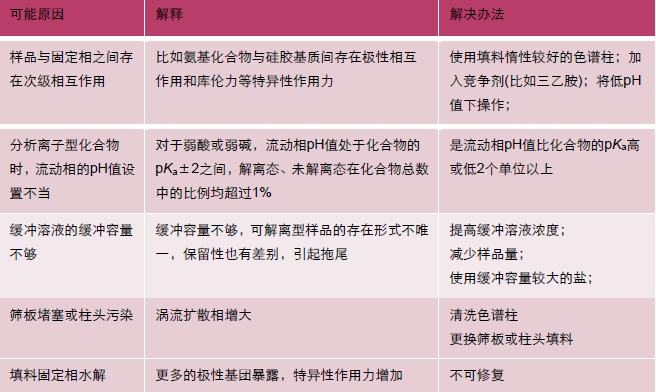
| 问题 | 现象 |
| A 基线漂移 |  |
| B 基线波动 | 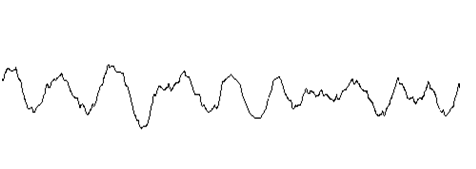 |
| C 宽大色谱峰 | 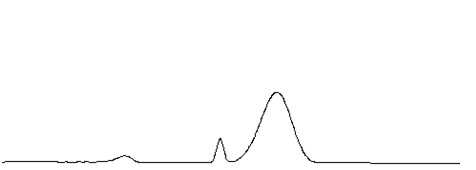 |
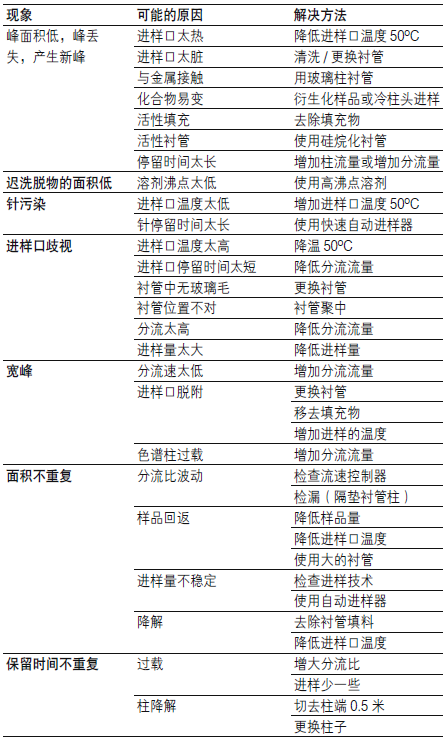
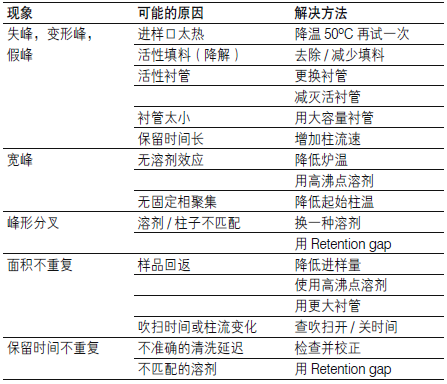
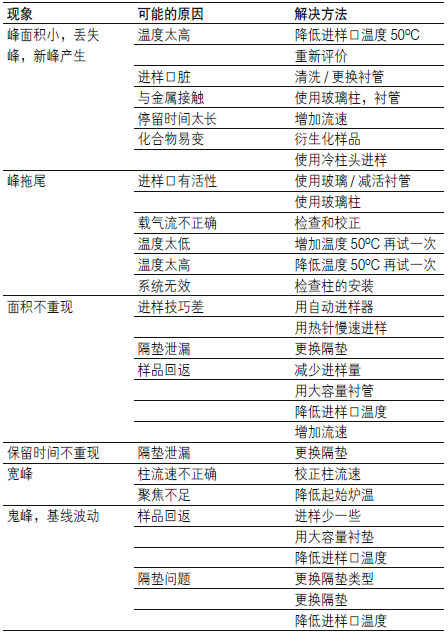
| 问题 | 现象 |
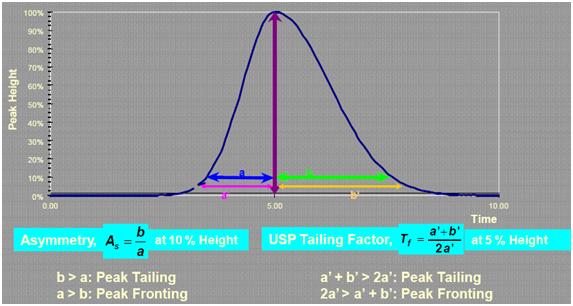
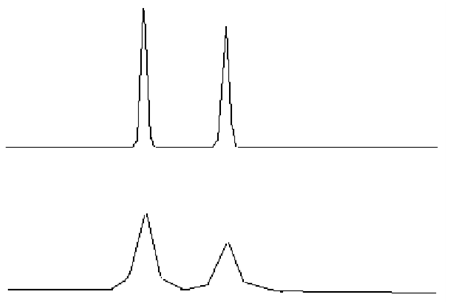
| A 正常峰形 | .bmp) |
| B 峰前延 | 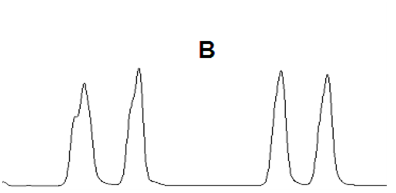 |
| C 峰拖尾 | 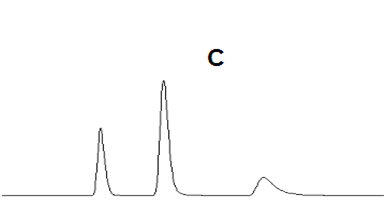 |
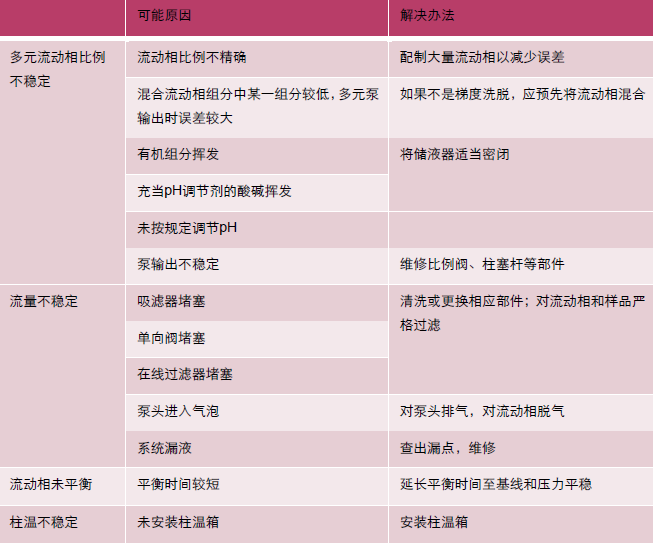
| 可能原因 | 改进办法 |
| 上样前,柱床干涸 | 重新活化并平衡 |
| 吸附剂填装松散,溶液出现短路 | 提出换货,或用小棍挤压柱床 |
| 样品液流速过快 | 降低流速,通常来说流速低于5 mL/min 时,结果是稳定的 |
| 可能原因 | 改进办法 |
| 样品溶液含有较多颗粒杂质,堵塞了筛板 | 上样前对样品溶液进行离心或过滤 |
| 样品溶液黏度太高 | 用合适的溶剂对样品进行稀释 |
| SPE 柱柱床太高,对液体的阻力过大 | 使用固相萃取仪,并在液体路出口减压 在液体入口加压 |