维权声明:本文为jncxyy2012原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
高效液相色谱法测定有关物质的方法建立的过程 有关物质系药品中除主成分以外的杂质,有关物质来源的两种途径:1.起始原料及合成过程中的中间体和副产物;2.储存过程中产生的降解产物。
由于有关物质的含量较少,所以选择专属性强、灵敏度高、重现性好的检测方法至关重要。目前常用的方法是高效
液相色谱法。现主要阐述高效
液相色谱法的建立过程。
高效
液相色谱法检测有关物质的方法有:(1)杂质对照品法(适用于已知杂质);(2)主成分自身对照法(紫外检测器),采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近)。
一、色谱条件的确定
1、查阅文献,看看是相关的资料,有的话可以参考。
2、没有文献,进行以下工作。
1)分析结构,看看有无紫外吸收,有就选用紫外检测器,没有的话可以选用蒸发光
散射检测器等。
2)分析结构,看看选用何种色谱方式进行分离(正相或者反相色谱)。
3)分析结构,看看流动相该如何选择,要不要选用一些离子对试剂。
4)与同类产品进行比较。
3、紫外扫描一下,找到最大吸收波长。将能得到的中间体、副产物、分解产物、样品配成相同浓度,在紫外扫描
分光光度计上扫描一下,选择工艺中最容易带入或产生的杂质的最大吸收处的波长作为测定波长。要是有
二极管阵列检测器就不需要这一步了。
4、查找资料,看看类似的样品的流动相,以它为起始流动相进行选择,如果没有,那就找专业书,看看它是怎么选择流动相的。
5、通过专属性验证所选色谱条件能否将各杂质与被测物分离检出 。应按0.5%(w/w)被测物浓度的各杂质量添加至被测物中,模拟被测物中可能存在杂质的状态,即有少量(约0.5%)杂质存在时能否与被测物达到完全分离(分离度大于2.0),以验证系统适用性。
二、最低检测限
得出达到S/N=2或S/N=3时的样品药品浓度。
三、供试品溶液浓度
一般在样品不过载的情况下,适当设定得高些,以保证该浓度在任何试验条件下,均有足够的检测灵敏度;也可以根据峰面积确定(经验),面积七位数时9开头,八位数时1开头。
四、线性及精密度试验
按药典规定,即采用该杂质对照品 (外购或合成获得)以外标法准确测定。此时,与含量测定相似,应进行线性试验。通常将杂质线性验证范围0.05%~1.2%)。精密度试验是以杂质百分浓度为0.5%进样,相对标准偏差不大于2.0。
五、回收率试验
杂质的定量试验可向原料药或制剂中加入已知量杂质进行测定。将各杂质以0.4%、0.5%、0.6% (w/w)的浓度加至被测物溶液中,以验证所采用的色谱条件是否可分离检测相应的各杂质以及与被测物中已存在的杂质是否累加,并观测累加量的准确性。
六、强降解试验
强降解试验条件有酸、碱、氧化、强光、高温破坏等。破坏时不能过于剧烈,一般以产生10%~20%杂质的条件为宜(《化学药物杂质研究技术指导原则》,同时采用二极管阵列检测器(DAD)验证主峰峰纯度,验证物料平衡。
七、其他色谱参数的确定
流速的选择:主峰保留时间约在l0min左右,这样主峰不易出现拖尾、堆积的现象,运行时间也不会太长。柱温的选择:不应超过35℃,既可增强被测物与杂质的分离度,也可避免因温度过高使主成分降解,导致测定误差。
溶剂的选择:首选流动相作溶剂为宜,以排除溶剂峰的干扰。也可以先用和流动相同比例有机溶剂溶解,再加入同比例水相。难溶物质,可采用甲醇或乙腈作溶剂以提高溶解性。
色谱图记录时间的设定:根据强力破坏试验和样品稳定性试验来规定色谱图记录时间,应能洗脱出有可能存在的全部杂质和经强力破坏试验产生的杂质,并规定运行时间主成分保留时间的几倍。
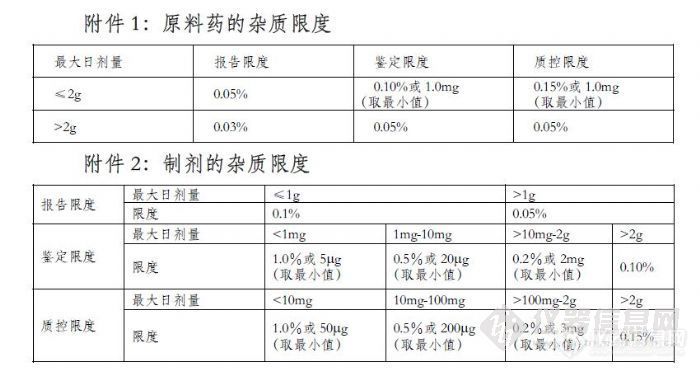
八、杂质限度的确定
杂质限度的制订应考虑如下因素:杂质及含一定限量杂质的药品的毒理学研究结果;给药途径;每日剂量;给药人群;杂质药理学可能的研究结果;原料药的来源;治疗周期;在保证安全有效的前提下,药品生产企业对生产高质量药品所需成本和消费者对药品价格的承受力。严格按照《化学药物杂质研究技术指导原则》来制定。