话不多少,直接进正题。
我做氨基甲酸乙酯,参考的是最新国标方法,一克样品,不含氯化钠的加氯化钠至饱和,加入d5-氨基甲酸乙酯同位素内标,混匀后上硅藻土小柱(安X,安XX,艾XX三个品牌的柱子我都用过,感觉效果差不多。艾XX的柱子流的很顺,样品做的很快,但是对实验会不会有影响,暂时不知道)。上柱吸附10分钟后,负压抽一会(不抽的话后面过柱感觉会很慢)。之后用10mL正己烷淋洗除杂,再用乙酸乙酯:乙醚(1:9)10mL洗脱(早期文献一般用二氯甲烷,但是能少一点毒性就少一点毒性嘛,还是用乙酸乙酯乙醚混合溶剂好一些。两者比例我试过2:8,1:9,0.5:9.5,效果差不多)。洗脱后氮气吹至近干,这一步最最头疼,要守着,万一全吹干了,基本回收就很低了。用同位素内标,可以吹到一毫升左右直接拿去上机。仪器用的是7000C的三杆,MRM模式,选取了62->45,62->44,74->44,62->62四个离子对。
气相柱用的DB-INNOWAX,调整升温程序,样品在14min出峰。
但是,问题来了
![]()
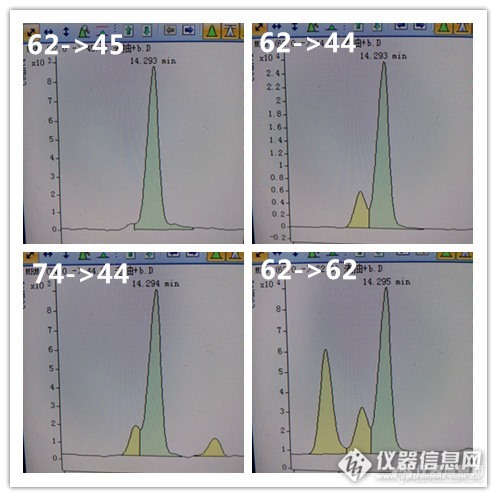
这是醋的四个离子对谱图,62->45这对离子还是比较干净,但是别的几对离子完全被干扰了,通过改变色谱条件,也没能将这三个峰分开。该样品含量大概是40ppb。
![]()
这个是酱油的四对离子,感觉完全无法定性定量,勉强积分,含量大概7ppb。
![]()
上面的酱油加标,加入后浓度是160ppb,后三个图还是可以看到明显的干扰。特别是62->62的离子对(相当于SIM模式),干扰非常明显。
![]()
这是总离子流图,总是这个分不开的三连峰,其中最右的是氨基甲酸乙酯。
国标给出的定量限是5ppb,我自己做的,感觉这么大的干扰,50ppb都难定准确,而且我还是用MRM做的。不知道我是前处理做的有问题,还是色谱条件不对,希望各位说一下你们做氨基甲酸乙酯的情况,一起探讨。
PS.
我最近发现酱油、醋、酸奶样品,加入氨基甲酸乙酯同位素内标后,用1mL乙酸乙酯涡旋提取一次,离心取上清液即可分析。内标的绝对回收率60%以上,总离子流图与过柱处理的基本相同。这种方法不知能否进一步改进来替代国标方法,大家可以试试。
![]()
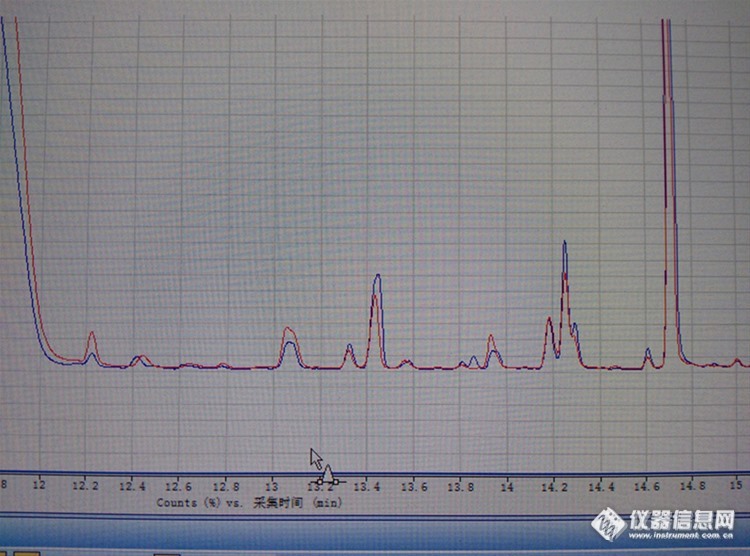
醋的总离子流图,红色的是硅藻土小柱提取,蓝色的是乙酸乙酯直接提取。
![]()
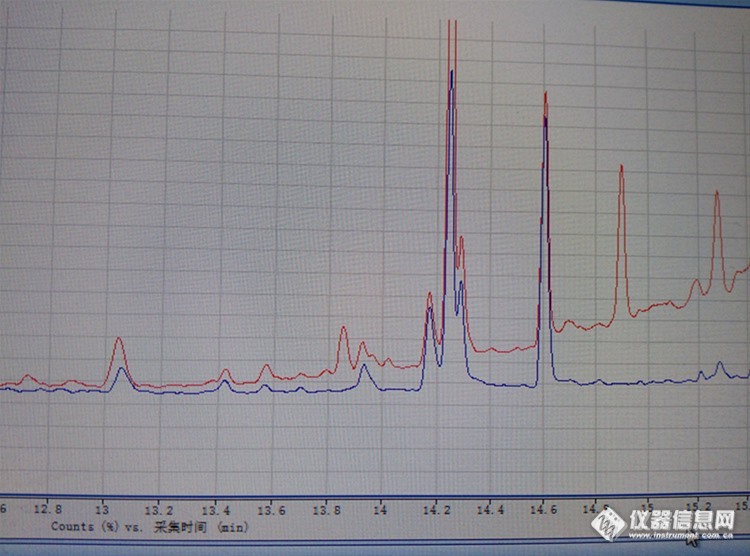
酱油的总离子流图,红色的是硅藻土小柱提取,蓝色的是乙酸乙酯直接提取。