维权声明:本文为v2696565原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
最近做的一个实验,刚刚写完,未发表,在投稿之中。前言
碱性橙2、碱性橙21、碱性橙22 和碱性嫩黄O都属于偶氮工业染料,主要用于腈纶、纺织品、纸张、皮革、家具等的染色
。这些染料具有致癌、致畸作用,对人体健康具有严重的危害。《中华人民共和国食品添加剂使用卫生标准》及《中华人民共和国食品卫生法》规定,碱性橙禁止作为食品添加剂。由于这些染料在中性及偏碱性的条件下,与蛋白质吸附牢固,不易褪色,而且成本低廉,因此一些不法食品生产商使用这些染料对豆制品进行染色,使之色泽光亮,欺骗消费者
。
目前,豆制品中这几种碱性染料的检测方法有高效
液相色谱法
、
液相色谱-质谱法
、薄层色谱扫描法等,尚未见高效毛细管电泳电泳法同时测定这几种碱性染料的文献报道。高效毛细管电泳法作为一种快速、低廉、绿色环保的分析方法,在分析领域应用很广泛。本研究采用乙酸铵水溶液提取、固相萃取浓缩净化的前处理方法,建立了碱性橙2、碱性橙21、碱性橙22 和碱性嫩黄O的毛细管区带电泳分析法,并应用与豆制品中的碱性染料的测定,方法准确可靠,简便快捷。
1 实验部分
1.1仪器与试剂
P/ACE MDQ型毛细管电泳仪(美国贝克曼库尔特有限公司),配有紫外检测器;未涂层熔融石英毛细管柱(40 cm,有效长度30 cm,内径75 μm,河北永年光导纤维厂); Turbo Vap自动浓缩仪(德国Biotage公司)。
碱性橙2 (99.6%)、碱性橙21(90.2%)、碱性橙22(95.4%)、碱性嫩黄O (79.0%)标准品(Dr Ehrenstorfer GmbH公司,德国); Oasis HLB固相萃取柱(美国waters公司);甲醇(HPLC),乙腈(HPLC),氨水(HPLC),乙酸铵(AR),磷酸二氢钾(AR);实验用水为超纯水(18.2 MΩ·cm)。
1.2 电泳条件
分离电压:25kV,分离温度:30℃,检测波长:210 nm,进样方式:压力进样(20psi,0.5s),运行缓冲溶液:40mmol/L磷酸二氢钾-20%乙腈(pH=2.5)。每次进样前按水、甲醇、水的顺序清洗毛细管各1min,然后用运行缓冲液冲洗2min。
1.3 样品前处理
1.3.1样品的提取
固体样品:将样品粉粹混匀,称取2 g(准确至0.001g)样品,置于10 mL具塞离心管中,加入5.00 mL提取液(50 mmol/L乙酸铵水溶液),超声提取30 min,3000 r/min离心10 min,将上清取出置于离心管中,再加入5.00 mL提取液提取一次,合并提取液后3000 r/min离心10 min后收集上清液,待过柱浓缩净化。
含油样品:将样品粉粹混匀,称取2 g(准确至0.001g)样品,置于10 mL具塞离心管中,加入5.00 mL提取液(50 mmol/L乙酸铵水溶液),超声提取30 min,3000 r/min离心10 min,将上清取出置于离心管中,再加入5.00 mL提取液提取一次,合并上清液后加入2 mL正己烷,涡旋振荡并萃取,弃去正己烷层,重复三次,待过柱浓缩净化。
1.3.2 样品的净化
分别以3 mL甲醇、3 mL水活化HLB固相萃取小柱,将上清液5mL(氨水调至近中性)加入萃取柱中,3mL的水淋洗后,加入2 mL 氨水-乙腈(10:90,v/v)洗脱,收集洗脱液。上样和洗脱过程流速控制低于1 mL/min。将洗脱液于30℃氮气吹至干,准确加入1.00 mL乙腈水(25:75,v/v),漩涡混匀30 s后过0.22 μm滤膜后待测。
2 结果与讨论
2.1 检测波长的选择
将碱性橙2、碱性橙21、碱性22、碱性橙O的单标储备液分别用超纯水稀释成50 μg/mL。以超纯水为空白,分别在190-800 nm范围对各物质的单标应用液进行紫外可见吸收光谱扫描,这四种碱性染料在450 nm和210 nm处均有较强吸收。由于食品样品基体复杂,在可见区域有较大干扰,并且由于仪器限制,不能在450nm处进行检测,因此选择210 nm作为实验的检测波长。
2.2 电泳条件的选择
2.2.1 缓冲试剂和缓冲液pH的选择
缓冲试剂的选择主要由所需的pH值决定,而该pH值又因样品的性质和分离效率不同而不同
。在毛细管区带电泳中,所需的待测物质在离子状态下才能实现分离。这四种碱性染料均有氨基基团,在酸性条件下可解离,因此选择磷酸、磷酸二氢钾作为分析的缓冲试剂。
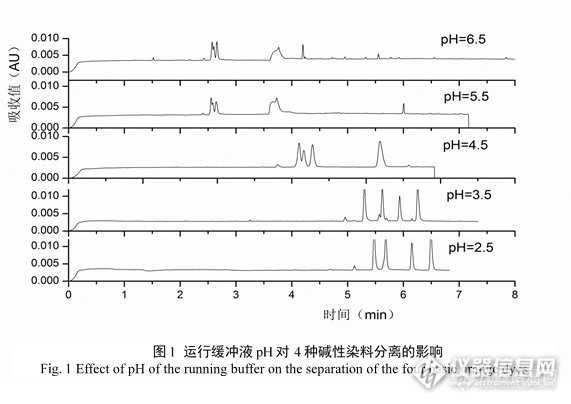
缓冲溶液的pH不仅能控制分析物的有效淌度,还控制着电渗流,同时对分析物的离子化程度有较大影响,因此是优化分离条件的重要因素。考察了40mmol/L磷酸二氢钾缓冲溶液在不同pH( 2.5、3.5、4.5、5.5、6.5)时对分离效果的影响。由图1可见,随着缓冲液pH值降低,出峰时间、峰高和峰面积均增加。在实验范围内,pH值越低,4种碱性染料的分离越好。故最终选择缓冲液的pH为2.5。
![]()
.2.2 缓冲液浓度的选择
缓冲溶液浓度通过影响溶液的粘度和电渗流,进而影响样品的分离情况。一般情况下,缓冲盐的浓度越高,电渗流越低,迁移时间就会延长,分离度增加。但是,缓冲盐浓度的升高却会增加焦耳热,使峰形变宽,峰面积下降。实验分别考察了浓度20 mmol/L、40 mmol/L、60 mmol/L、80 mmol/L 磷酸二氢钾盐浓度的条件下时,被测的这4种碱性染料的迁移时间和分离度。随着磷酸二氢钾浓度的增加,四种碱性染料分离度增加,但电流同时增加,迁移时间延长,因此综合考虑,选择40 mmol/L为最佳的磷酸二氢钾浓度。
2.2.3 添加剂的种类和浓度
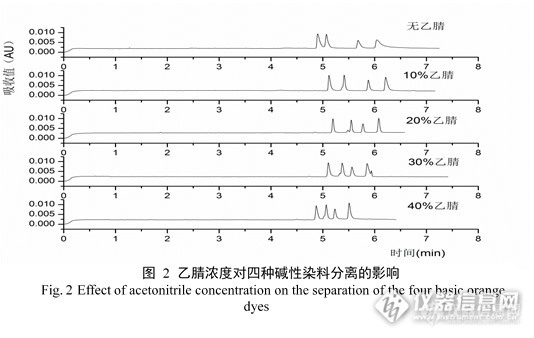
添加有机溶剂,可以减少毛细管电泳的电渗流,降低焦耳热的产生,从而有利于物质的分离。有机添加剂通常选择其有较高的偶极矩和介电常数,并对被测物质有良好的溶解性。一般来说,甲醇、乙腈是比较合适的有机添加剂。本实验对比了不同有机溶剂(甲醇、乙醇、乙腈、丙酮)对四种碱性染料的分离的影响。实验结果表明,添加甲醇、乙醇、丙酮的添加使峰形拖尾,同时迁移时间也明显增加,而添加乙腈能够明显改善峰形,增加分离度。因此本文进一步比较了不同乙腈比例对四种碱性橙分离的影响(图2)。实验结果表明,随着乙腈比例的增加,峰形明显改善,分离度增加,迁移时间减少。但过多的乙腈会容易在毛细管内形成气泡,造成基线波动,因此在综合考虑分离效果和基线波动等因素,最终选择添加20%的乙腈。
![]()
2.2.4 分离电压和温度
实验比较了分离电压15-30kV范围内时对四种碱性染料的分离效果的影响。随着分离电压的升高,迁移时间缩短,但基线噪声随之增大。最终选择了20kV作为分离电压。
毛细管的柱温对迁移时间、电渗流、测量重现性都有一定的影响
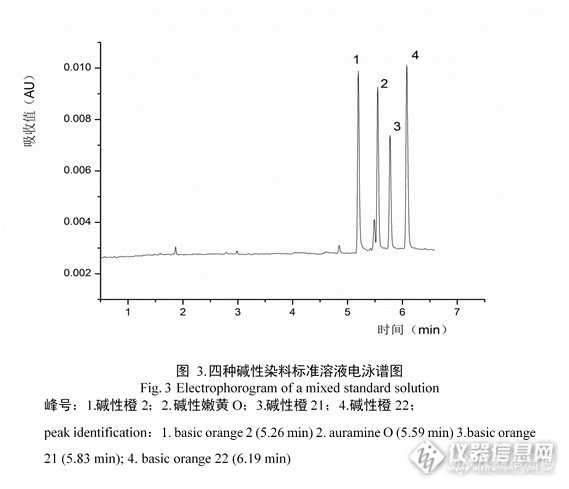
。本实验分别比较了分离温度为20℃、25℃、30℃、35℃下四种碱性染料的分离情况,在分离温度为30℃时,四种碱性染料有较好的分离度并且迁移时间较短。优化好的标准色谱图见图3。
![]()
2.3 样品前处理条件的选择
2.3.1 提取液的选择
文献
中报道用于提取这四种碱性染料的溶液有乙酸-甲醇(10:90,v/v)、氨水-甲醇(10:90,v/v)、乙醇、乙醇水溶液(30:70,v/v)、乙酸铵水溶液等。本文以空白豆腐皮为样品进行加标,考察了这些溶液对这四种碱性染料的提取效果。实验结果表明,50 mmol/L的乙酸铵水溶液回收率最高,同时便于下一步的浓缩净化过程,故选择50 mmol/L的乙酸铵水溶液作为提取溶液。
2.3.2 固相萃取柱的选择
在碱性染料净化的固相萃取柱的选择上,HLB柱
、C
18柱
、MCX柱
均有报道本实验考察了这三种固相萃取柱的净化效果。结果表明,MCX固相萃取柱与碱性橙的结合能力很强,难以洗脱,必须使用二氯甲烷、三氯甲烷等溶剂才能洗脱,HLB固相萃取柱在杂质的去除和回收率上均优于C
18固相萃取柱。因此选择HLB作为净化固相萃取柱。
2.3.4洗脱溶剂的选择
实验考察了不同类型洗脱剂(氨水甲醇溶液、氨水乙腈溶液、氨水丙酮溶液)的洗脱效果,结果表明,氨水甲醇的洗脱能力差,不能够将碱性嫩黄O完全洗脱下来,而氨水丙酮洗脱能力太强,洗脱杂质多,不利于检测,而氨水乙腈在保证将这四种碱性染料洗脱下来的前提下,洗脱的杂质少,最终选择氨水乙腈作为洗脱溶剂。
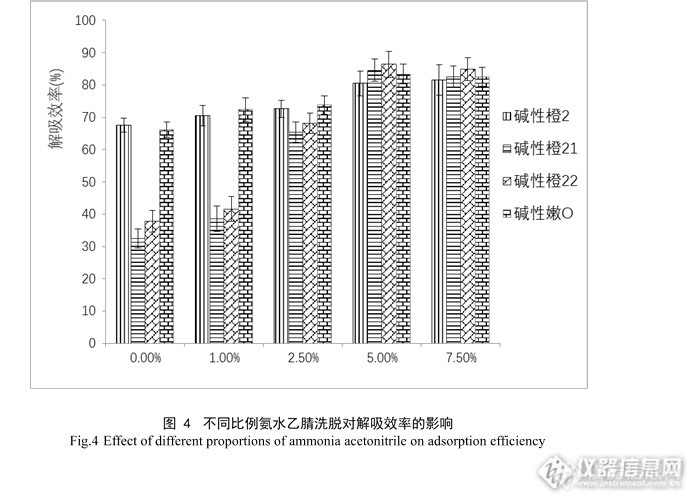
进一步比较了不同浓度氨水-乙腈(1.0%、2.5%、5.0%、7.5%,v/v)洗脱能力,从图4可见,随着氨水比例的增加,碱性橙21、碱性橙22的回收率有明显上升。当到达5%时,这四种碱性染料的解析效率达到最高,最终选择5%氨水乙腈作为洗脱溶剂。
![]()
2.4 方法性能考察
2.4.1 回归方程、线性范围、检出限和定量限
分别取标准储备液适量,配制5个浓度水平的标准系列,在优化的电泳条件下,由低浓度到高浓度分别进样测定,从而得到各物质的回归方程以及线性相关系数。回归方程中
y表示峰面积(AU•min),
x表示浓度(mg/L)。根据3倍信噪比和10倍信噪比对应的样品中待测物质含量,确定方法检测限和定量限,结果分别0.5-0.7mg/kg和1.7-2.3mg/kg。
表1方法的线性范围、回归方程、检出限和定量限 Tab. 1 Linear ranges, correlation coefficients, limit of detection, limits of quantification of the method |
检测物质 | 线性范围
ρ/(mg/L) | 回归方程 | 相关系数 | 检出限 ω/(mg/kg) | 定量限 ω/(mg/kg) |
碱性橙2 | 1.25-50 | Y=1830.4X-37.56 | 0.9988 | 0.5 | 1.7 |
碱性嫩黄O | 1.25-50 | Y=1744.8X-901.85 | 0.9978 | 0.6 | 2.0 |
碱性橙21 | 1.25-50 | Y=1286.2X-600.96 | 0.9977 | 0.7 | 2.3 |
碱性橙22 | 1.25-50 | Y=2115.1X-956.22 | 0.9979 | 0.5 | 1.7 |
2.4.2 精密度实验
取浓度为25 μg/ml的4种碱性染料的混合标准溶液,按本法进行测定,在一天内连续测定6次,以峰面积的相对标准偏差(relative standard derivation,RSD)考察方法的日内精密度。连续6天进行测定,考察方法的日间精密度。由表2可见,这4种碱性染料的日内精密度(RSD)为1.55%-3.55%,日间精密度(RSD)为2.48%-10.58%。
表2方法的日内和日间精密度(n=6) Tab. 2 The intra- and inter-day precision of the method(n=6) |
| 检测物质 | 日内精密度 | | 日间精密度 |
测定范围 ρ/(mg/L) | 测定均值 ρ/(mg/L) | RSD /% | | 测定范围 ρ/(mg/L) | 测定均值 ρ/(mg/L) | RSD /% |
碱性橙2 | 23.8-25.8 | 24.8 | 3.05 | | 21.6-26.7 | 24.7 | 9.58 |
碱性嫩黄O | 24.2-25.9 | 24.8 | 2.43 | | 23.5-27.9 | 25.4 | 6.57 |
碱性橙21 | 24.8-26.0 | 25.4 | 1.79 | | 23.8-26.8 | 25.3 | 4.36 |
碱性橙22 | 25.1-26.3 | 25.6 | 1.55 | | 24.5-25.8 | 25.4 | 2.48 |
2.4.3回收率实验
取12份平行样品,其中3份测定本底值,其余分别加入3个不同水平的混合标准溶液,每个水平测定三个平行样品,进行加标回收实验,并计算峰面积的RSD值来考察样品溶液的精密度。实验结果(表3)表明,本法的加标回收率范围为73.6%-93.1%,加标样品的RSD范围为1.28%-4.69%,能够满足分析测定的要求。
表3.方法的加标回收率(n=3) Tab. 3. The spiked recoveries of the method (n=3) |
| 检测物质 | 本底值 | 加入量 | 测量值 | 回收率 /% |
RSD
/% |
| ω/(mg/kg) |
| 碱性橙2 | ND |
10.0 |
7.85 | 78.5 | 3.56 |
40.0 |
33.7 | 84.3 | 2.53 |
80.0 |
74.5 | 93.1 | 1.96 |
| 碱性嫩黄O | ND |
10.0 |
7.92 | 79.2 | 4.14 |
40.0 |
32.6 | 81.5 | 2.56 |
80.0 |
68.5 | 85.6 | 3.65 |
| 碱性橙21 | ND |
10.0 |
8.12 | 81.2 | 4.69 |
40.0 |
34.1 | 85.3 | 2.47 |
80.0 |
70.3 | 87.9 | 1.32 |
| 碱性橙22 | ND |
10.0 |
7.36 | 73.6 | 3.28 |
40.0 |
31.2 | 78.0 | 1.56 |
80.0 |
69.4 | 86.8 | 1.28 |
ND表示未检出
2.5方法应用
应用本法分析检测了腐竹和豆腐干共9种豆制品中的碱性染料,结果均未检出。图5为不同种类样品及其加标后的电泳图。
![]() 3小结
3小结本实验对毛细管电泳的仪器条件(分离电压和分离温度)和缓冲液条件(pH、浓度、添加剂等)进行了优化,在6.5min内实现了对四种碱性染料的快速分离。对样品前处理的条件(提取条件、固相萃取条件)进行了优化,结果表明,方法精度度和准确度能够满足食品测定的要求。对实际样品进行了测定,结果表明本方法简单、准确、快速,适用于食品样品碱性染料的分析。