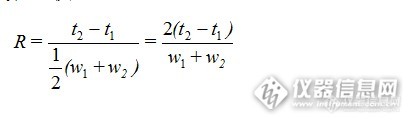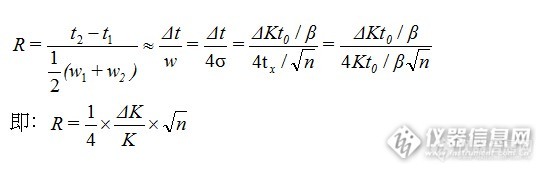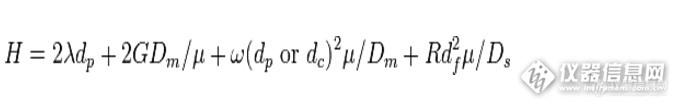在
气相色谱的检测中我们经常会遇到色谱图里出现一些非目标物质峰,或者多次进样后峰形有变化、分离度下降等问题,今天就和大家聊聊怎么赶走这
气相色谱中的“不速之客”。
首先,咱们来聊一聊鬼峰 鬼峰可能来自隔垫流失、载气杂质及被污染的气路管线;其次可能来自载气中微量氧气、水或其它物质等与固定相的反应产物;以及前次进样的高沸点残留组分流出,污染的进样口和柱头都可能导致鬼峰产生。如:
①载气不纯,杂质在低温时凝聚,当温度升高时就会流出;
②前次进样残留的高沸点组分峰流出;
③液体样品中的空气峰;
④样品使色谱柱上以前吸附的杂质解吸出来;
⑤样品在进样口或色谱柱中高温下分解;
⑥样品被污染;
⑦高温下或程序升温时进样口隔垫分解;
⑧样品与固定液或载体相互作用产生杂质;
⑨玻璃棉或进样器带入的污染物。
解决方案①空白运行时出现鬼峰
通常在程序升温过程中出现,是由于在相对较低的起始温度下,柱头会聚集污染物,随着柱温升高被释放并在谱图上表现出来造成。
空白运行或进溶剂时出现鬼峰,可能是之前进样残留的高沸点组分流出所致,这时多进样几次空针或进溶剂运行,鬼峰就可以消失。一段时间内色谱柱都处于初始温度下时,后续进样常常会出现鬼峰,例如,每天或每周的最初几次运行经常会出现鬼峰。
污染的进样口也可能导致鬼峰,因为样品挥发或热解以后会被吹扫进入色谱柱,可尝试降低进样口温度,如果能消除或减弱鬼峰,则是进样口被污染导致,需要清洗进样口,更换衬管、密封圈和隔垫等。
②纯样品进样时出现鬼峰
当纯样品进样时出现鬼峰,可先只用溶剂做一次空白运行,如果鬼峰仍然出现,那么鬼峰与该样品无关。假设样品是纯净的,出现鬼峰的一个常见原因是进样口过热造成组分分解,这个问题可以通过降低进样口温度来确认。
如果运行一次空白,原有的鬼峰没有出现,则说明该样品受到污染,需要检查样品处理过程中潜在污染来源,如样品提取、纯化、转移和存储等环节。
在降低进样口温度、减慢气化速度不引起色谱峰展宽的前提下,应尽可能使用低的进样口温度来操作,避免出现高温导致分解的情况;或应使用挥发性更强的溶剂,极端情况下,分析之前应对可能分解的样品先进行衍生化,降低进样口带来的变异。
金属柱也可能导致样品降解。此时,鬼峰往往比它们附近的峰要宽,因为这些鬼峰成分是在通过整个柱长的过程中产生的。如果确认这是产生鬼峰的原因,就有必要换成玻璃柱或熔融石英柱。
空白样品测定时怎么出现待测成分了? 导致空白样品测定时出现待测成分色谱峰的主要原因,可分为以下几种:
①可能是由于制备空白的试剂含有待测组分,如试剂被污染,或装空白试剂的容器被污染。
②可能是仪器之前测试过同样的样品,之后系统内有测试样品的残留。这主要和温控系统、柱系统和检测系统的设置和配置有关,如设置温度不当时,上一次样品组分在仪器内会有残留,干扰后面的测定。
③ 仪器的气体、管路、气体净化器、进样口等被污染,有时空白运行时也会出现待测成分。
④ 空白样品中含性质与所测样品极相似的成分,误以为空白样品中有待测成分,如该成分保留
时间与待测样品相同等。
解决方案 空白样品溶液出现待测成分色谱峰,应按下列方法逐一检查:
首先,考虑制备用的试剂、制备过程中的样品以及盛装容器是否被污染。分析待测组分含量低的样品时,空白运行时出现的待测成分杂质会严重干扰实验结果。如果是这些原因造成的,清洗容器,重新制备,有问题的试剂需更换。
其次,要检查温控系统、柱系统和检测系统,如温度设置是否合适,选用的色谱柱是否合适等,如有问题需改变温度条件或更换色谱柱。
如果使用上面的方法仍没有解决问题,应考虑仪器的气体、管路、气体净化器、进样口等是否被污染。如要检查气体发生器的碱液有没有漏液,是否需要更换,以及气体钢瓶载气的纯度是否达到要求;检查气体净化器的分子筛、硅胶等是否需要更换或活化;同时,也要检查进样系统,如进样口隔垫、衬管、分流出口管路等是否被污染。
如果经过上面的排除方法,仍没有解决问题,那么得考虑是否是由于空白样品中含性质与所测样品极相似的成分造成的,可选择多种定性方法组合定性判断,如相对保留值定性、双柱和多柱结合等定性方法。
空白运行时出现待测成分,会影响实验结果的判断。例如,试验新方法时,一般先按照标准方法设置柱温升温程序,如实验时一个样品分析需要60min,为减少分析时间而提高升温速率,虽然分析时间减少了,但由于与固定相接触的时间也减少了,可能导致进下一个样时,在待测成分保留时间位置出现上一个样的杂质,干扰判断。另一种情况,由于温度设置不当,高沸点的组分没完全流出,而在下一次进样测定时才流出,而保留时间又刚好一致,会误导误判断。
分离度下降了?! 色谱分离度又称分辨率,用来表征两个相邻色谱峰的分离程度,用R表示。R等于相邻色谱峰保留时间之差与两色谱峰峰宽均值之比。计算公式为:
![]()
R越大,表明相邻两组分分离越好。一般说当R<1时,两峰有部分重叠;当R=1.0时,分离度可达98%;当R=1.5时,分离度可达99.7%。通常用R=1.5作为相邻两组分已完全分离的标志。当R=1时,称为4σ分离,两峰基本分离,裸露峰面积为95.4%,内侧峰基重叠约2%。R=1.5时,称为6σ分离,裸露峰面积为99.7%。R≥1.5称为完全分离。《中国药典》规定R应大于1.5。为了判断分离物质对色谱柱在色谱柱中的分离情况,常用分离度作为柱的总分离效能指标,
从塔板理论可以推论出:
![]()
从这里我们可以清晰地看到影响分离度R的因素只有三个,即:两临近组分的分配系数(容量因子)之差ΔK、平均分配系数K、柱理论塔板数n。因此分离度R与柱长L(或柱理论塔板数n)的平方根成正比,与分配系数之差ΔK成正比,与平均分配系数K成反比。分配系数与固定相的性质、厚度及柱温有关:①与样品极性相似的固定相,分配系数大;②固定液液膜厚度增加,分配系数增大;③温度降低,分配系数增加。
因此我们仅从塔板理论来看,载气流速并不影响分离度R的。我们知道,这并不符合我们的实际经验。事实上,分离度R与载气流速是密切相关的。为什么会出现这个情况呢?这是因为塔板理论的四个假设并不成立,存在偏差。为了修正这些偏差,1956年荷兰人Van Deemter提出了速率理论
。
速率理论是对塔板理论的一个修正。范第姆特方程(Van Deemter equation)在色谱学中是综合考虑了分离过程中引起峰展宽的物理因素、动力学因素和热力学因素后得到的单位柱长的总峰展宽与流动相流速的关系式。
一般来说,影响峰展宽的因素包括多路径效应,扩散(纵向和横向)与固定相和流动相间的传质阻力。通常用理论塔板高度来表示色谱分离过程中的峰展宽。范德姆特方程呈双曲形函数的形式,表明流动相的流速存在一个最优值,在该点柱效最高。范第姆特方程最常用的形式如下式所示,该式直观地反映了流动相流速对于分离的影响。
![]()
式中H 为理论塔板高度,A, B,C为常数,μ表示流动相的流速。
A项反映的是被分析物在填充柱中可能采取不同的路径,因而经过的路程也不一样长,引起色谱峰的展宽,这就是“多路径效应”。在毛细管开管柱中不存在多路径效应,这一项为零。
B/μ表示的是因为色谱柱各部分存在浓差而引起的纵向扩散带来的峰展宽。
Cμ表示达到固定相与流动相的平衡之后由于在固定相与流动相传质存在着阻力引起的峰展宽。
范第姆特方程的完整形式如下:
![]()
式中:
λ 是一个与柱内填料粒度均一性与填充状态有关的常数;
dp 为填料的粒径;
G、ω和R 为常数;
Dm 是被分析物在流动相中的分子扩散系数,与载气的性质和柱温有关;
dc 是毛细管的直径;
df 是固定相的膜厚度;
Ds 是被分析物在固定相中的分子扩散系数,与固定相的性质有关。
由以上相关理论可以推论出,色谱柱的分离度与色谱柱的长度、固定相的性质、液膜厚度、载气流速和性质、柱温均有关系。
①分离度R与柱长L的平方根成正比,因此,采用较长的色谱柱可以获得更高的分离度,但相应的分析时间也会延长。而且色谱柱越长压降越大,柱过长会增大进出口压力比,相反会降低分离度。
②固定相的性质与分析物越相似,分离度越高;
③从上述公式中可以看出,液膜厚度的增加会导致塔板高度的增加,柱理论塔板数降低,同时还会增加分析物在固定相中的分配系数,理应导致分离度降低,但事实上,在分配系数增加的同时,ΔK增加更为显著,所以液膜厚度的增加能提高分离度;
④分子质量大的载气(N
2、Ar)的优点是扩散作用小,缺点是在柱中压降大、流速慢,即分析周期长。分子量较小的载气(H
2、He)传质阻力小,有利于提高分析速度,但浓度较高的介质易在其间形成扩散,影响分离度,所以在实际测量中H
2、He气体一般都用于介质浓度较低的区域并提高其流速,减少扩散的影响。
⑤色谱柱存在一个最佳载气流速,但实际分析中为缩短分析时间,流速通常稍大于最佳流速。
⑥提高柱温能提高分析物在流动相和固定相中的扩散系数,降低传质阻力,加快分析速度,同时使色谱峰变窄变高,但会导致分离度的下降。
⑦此外,色谱分离是样品在固定相和流动相中反复分配的过程,过载后本来该进入固定相的样品在固定相上得不到保留(例如:本来在这点有50%的样品能保留在固定相上,现在只能保留30%,因为固定相上样品饱和了),就被流动相推着往前走,因此造成了保留变化(主要是分配系数发生了改变),因此进样量与分离度也是有关系的。
因此,引起分离度降低的原因可能有:a.载气流速设置不当;b.柱温设置不当;c.色谱柱受到污染,柱效下降;d.进样量太大;e.样品的浓度发生改变。
解决方案①载气流速设置不合理,可以通过降低载气流速来提高分离度;
②柱温设置不当,降低柱温,或改用程序升温;
③色谱柱受污染,应清洗柱子;
④减少进样量,提高进样技术;
⑤提高气化室温度;
⑥减少系统的死体积,主要是色谱柱要连接到位,毛细管色谱柱要分流,选择合适的分流比。